|
Shivananda
Premalatha R.
Raheela
Gayathri P.
From the Department of Pediatrics, Vani Vilas
Children's Hospital, Bangalore, India.
Reprint requests: Dr. Shivananda, Professor and Head, Department of
Pediatrics, Vani Vilas
Children's Hospital Bangalore, India.
Manuscript Received: February 29, 1998; Initial review completed:
April 3, 1998;
Revision Accepted: June 28, 1998
Triple-A syndrome describes patients with achalasia,
alacrimia and glucocorticoid deficiency with normal mineralocorticoid
activity. This form of chronic adrenal insufficiency is characterized by
isolated deficiency of glucocorticoid, elevated levels of corticotropin
and normal aldosterone production. The salt losing manifestations of
most other forms of adrenal insufficiency do not occur, instead patients
present primarily with hypoglycemic seizures and pigmentation during the
first decade of life. Most patients exhibit achalasia of the bcardia,
deficient tear production and other autonomic dysfunction. We describe a
child with this syndrome in view of the paucity of documentation on this
subject in the Indian literature.
Case Report
A 9-year-old girl from Bangalore, the 3rdchild of a non-consanuinously
married couple, born after full term gestation by normal vaginal
delivery presented to us with complaints of difficulty in swallowing
mainly for solids and vomiting after taking food since 6 months and
muscle weakness and clumsiness since 3 months. Her postnatal and infancy
period was uneventful. Growth and development was normal. At the age of
3 years she developed blackish pigmentation of gums, lips and tongue,
gradually involving the whole body especially' over pressure points and
extensor aspects of phalangeal joints. At the age of five, she has
undergone tonsillectomy for recurrent attacks of fever and throat pain,
The post-operative period was uneventful. There was no history of
seizures or salt craving. Absence of tears since infancy was observed by
the mother.
Her first brother developed hyper pigmentation at the age of 2 years. He
also had deficient tear production since infancy. He expired at the age
of
3V:z
years following an episode of fever, vomiting and
convulsions (probably due to hypoglycemic attack). Her second
sibling-a-female child, had deficient tear production since infancy and
developed hyper pigmentation at the age' of 2 years. At the age of 5
years, the child developed an attack of convulsion and altered sensorium
and expired the same day. History of achalasia was absent in these two
children. Both parents were normal and there was no history of any
adrenal disorder, swallowing disorder or lack of lacrimation.
Physical examination revealed height of 126 cm (25th percentile) and
weight of 21 kg (5th percentile). Pulse rate was 90/min and BP was
100/70 mm of Hg without any evidence of postural hypotension. Hyper-
pigmentation was noticed on gums, lips, tongue, pressure points,
extensor aspects of phalangeal joints and over palmar creases. Tongue
papillae were normal. Numerous fine palmar creases were noticed.'
External genitalia were normal. Pupillary size was normal. Child had
nasal voice. Pharyngeal reflex was normal. Neurological examination
revealed brisk deep tendon reflexes, normal muscle tone with flexor
plantar response and clumsy gait. Schirmer test and tear film break-up
time we're indicative of deficiency of tear production in both the eyes.
Investigations revealed normal hemogram, urinalysis, renal functions,
blood sugar and serum electrolyte levels. ESR was 30 mm at the end of
one hour. Urine 17 ketosteroids was 5 mg/24h (normal 6-15 mg/24 h).
Cortisol level was low. How- ever, it returned to normal after treatment
(Table I). Mantoux test was negative. Plain X-ray of chest and
abdomen was normal. Abdominal ultrasound examination was also normal.
Barium swallow showed achalasia of cardia (Fig. 1).
The patient was given cortisol replacement therapy and methyl cellulose
eye drops. She was on prednisolone 5 mg daily to start with and
subsequently the dose was reduced to 2.5 mg on alternate day. With this
dose the serum cortisol levels were maintained within normal range. With
this treatment, pigmentation of the skin and oral cavity has decreased.
Nasal tone has come down and the child is taking food normally. Muscle
weakness has reduced.
TABLE I
Sem1ll Cortisol Levels (pg/dl)
Time of the day
|
Observed
(Range) |
After
treatment |
|
Morning |
0.2 (7-31) |
12.7 |
|
Evening |
0.5(4-14.5) |
10.8 |
Figures in parenthesis indicates normal values.
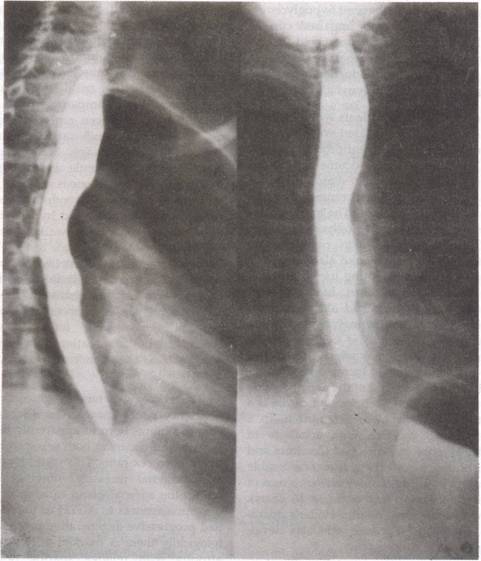 |
|
Fig. 1. Barium swallow showing achalasia of cardia. |
Discussion
The syndrome of familial glucocorticoid deficiency first described by Shepard et
al.(1) is characterized by the onset in
early childhood of recurrent hypoglycemia, skin pigmentation and impaired cortisol synthesis. Familial glucocorticoid deficiency associated with achalasia of cardia
and deficient tear production was described in 1978 by Allgrove et al.(2).
In a series of 20 patients with glucocorticoid deficiency associated with achalasia of cardia and deficient tear production, cortisol deficiency was documented in all cases(3). The age at which symptoms of adrenal insufficiency began ranged from 1.0 day to 8 years 3 months. The initial symptoms were related to hypoglycemia associated with increased skin pigmentation. Ninteen of the patients had alacrimia since early infancy necessitating use of artificial tears to avoid discomfort. In the present case also, the patient had alacrimia
since infancy. Evidence of hypoglycemia was not seen in the present case because
of the absence of severe illness which precipitates this metabolic event.
Increased skin pigmentation was noticed at the age of 3 years in the present case. Achalasia of cardia is a relatively rare problem in children. In a series of 35 children with achalasia of cardia(4), only one case had alacrimia and cortisol deficiency. There is a considerable variation in the ages at which symptoms of achalasia
develop (6 months to 16 years). Some of them may require Heller's operation. In the present case achalasia developed at the age of 8th years.
Skin changes commonly seen are hyperkeratosis of palms with multiple fine
fissures and cutisanserina, pigmentation of gums, lips, pressure points and extensor aspects of phalangeal joints. This girl had hyperpigmentation and fine palmar creases.
festations reported in this syndrome involve both central and peripheral neurons. The most common findings were hyper- reflexia, increased limb tone, extensor plantar response, muscle weakness and wasting. Dysarthria, nasal speech, ataxia, clumsiness, sensory impairment and optic atrophy have also been reproted. Usually the neurological manifestations are noted around 10 years of age. Many of these manifestations are subtle and picked on clinical examination if one is aware of these findings in Triple A syndrome. In the present case, the child had hyper-reflexia, nasal speech and clumsy walking suggestive of upper motor neurone
lesions. Even though some degree of intellectual impairment has been
reported(4), this girl did not have any intellectual impairment. The common
autonomic disturbances reported are postural hypotension, unequal pupils, absent
or reduced sweating, abnormal histamine test and abnormal methacholin test(4).
The etiology of familial glucocorticoid deficiency is not known. The disorder occurs in siblings born to apparently healthy parents and occurs in both sexes with relatively even sex ratio. This could be due to an autosomal recessive inherited defect within the adrenal gland causing primary unresponsiveness to ACTH or to an inherited
progressive degenerative process. Histologically there is marked adrenocorticol atrophy with relative sparing of Zona glomerulosa. It has been suggested that the unresponsiveness of adrenal cortex may be due to failure of membrane attachment or to failure of activation of adenyl cyclase by corticotropin. Abnormality of mineralocorticoid function has been described indicating
progressive degenerative process involving the adrenal gland(5).
Other manifestations of Triple-A Syndrome are neurological involvement and' The natural course of familial glucocor-autonomic disturbance. Neurological maniticoid
deficiency is not completely understood. Achalasia of the cardia may be the first symptom, other symptoms unfold as the age advances. Some of the patients may develop mineralocorticoid deficiency later. The present case may have a comparitively better prognosis because of the absence of hypoglycemia and the fact that her symptoms have reduced with treatment. It is worth assessing the adrenal function in any child who presents with achalasia especially if associated with alacrimia(6). Early recognition of familial glucocorticoid
deficiency will prevent hypoglycemic convulsions, neurological sequalae and death.
|
1.
Shepard TH, Landing BH, Mason DG. Familial Addison's Disease: Case Report of two sisters with corticoid deficiency unassociated with hypo-aldosteronism. Am
J
Dis Child 1959; 97: 154-162.
2.
Allgrove J, Clayden GS, Grant DB, Maccualey Je. Familial glucocorticoid deficiency with achalasia of the cardia and
deficient tear production. Lancet 1978; 1: 1284-1286.
3.
Grant DB, Barnes ND, Dumic M, Ginalska-Malinowaka M, MilIa PJ, Petrykowski WV, et al. Neurological and adrenal dysfunction in the adrenal insufficiency / alacrimia/ achalasia (3A) syndrome. Arch Dis Child 1993; 68: 779-782.
4.
Nihout-Fe 'Ke 'Te' C, Bawab F, Lortat-Jobb S, Arhan P, Pellerin P. Achalasia of
the esophagus in childhood: Surgical treatment in 35 cases with special reference to familial
cases
and glucocorticoid
association.
J
Pediatr Surgery 1989; 24:
1060-1063.
5.
Lanes R, Plotnick LP, Byrum TE, Lee P A, Casella JF, Fox CE, et al. Glucocorticoid and partial mineral corticoid deficiency associated with achalasia.
J
Endocrinology Metabolism 1980; 50: 268-270.
6.
Shah BR, Fiordal-isi-I, Shienbaum K, Fiberg L. Familial glucocorticoid in a girl with familial hypophosphatemic rickets. Am
J
Dis Child 1988; 142: 900-903.
|
