|
|
|
Indian Pediatr 2021;58:461-481 |
 |
Steroid Sensitive Nephrotic Syndrome:
Revised Guidelines
|
|
Aditi Sinha, 1 Arvind
Bagga,1 Sushmita Banerjee,2
Kirtisudha Mishra,3 Amarjeet Mehta,4 Indira Agarwal,5 Susan Uthup,6 Abhijeet Saha,7
Om Prakash Mishra8 and
Expert Group of Indian Society of Pediatric Nephrology*
From 1Division of Nephrology, Departments of Pediatrics, All India
Institute of Medical Sciences, New Delhi; 2Institute of Child Health,
Kolkata; 3Chacha Nehru Bal Chikitsalaya, Delhi; 4Sawai Man Singh Medical
College, Jaipur; 5Christian Medical College, Vellore; 6Trivandrum
Medical College, Thiruvananthapuram; 7Lady Hardinge Medical College, New
Delhi; 8Institute of Medical Sciences, Benaras Hindu University,
Varanasi, India. *List of expert group members provided in Annexure I.
Correspondence to: Dr. Arvind Bagga, Division of Nephrology,
Department of Pediatrics, All India Institute of Medical Sciences,
Ansari Nagar, New Delhi 110029, India.
Email: [email protected]
Published online: March 20, 2021;
PII: S097475591600301
|
Justification: Steroid sensitive nephrotic syndrome (SSNS) is one of
the most common chronic kidney diseases in children. These guidelines
update the existing Indian Society of Pediatric Nephrology
recommendations on its management. Objective: To frame revised
guidelines on diagnosis, evaluation, management and supportive care of
patients with the illness. Process: The guidelines combine
evidence-based recommendations and expert opinion. Formulation of key
questions was followed by review of literature and evaluation of
evidence by experts in two face-to-face meetings. Recommendations:
The initial statements provide advice for evaluation at onset and follow
up and indications for kidney biopsy. Subsequent statements provide
recommendations for management of the first episode of illness and of
disease relapses. Recommendations on the use of immunosuppressive
strategies in patients with frequent relapses and steroid dependence are
accompanied by suggestions for step-wise approach and plan of
monitoring. Guidance is also provided regarding the management of common
complications including edema, hypovolemia and serious infections.
Advice on immunization and transition of care is given. The revised
guideline is intended to improve the management and outcomes of patients
with SSNS, and provide directions for future research.
Keywords: Calcineurin inhibitors, Frequent relapses,
Levamisole, Minimal change nephrotic syndrome, Mycophenolate mofetil,
Rituximab, Steroid dependence.
|
|
N ephrotic syndrome,
characterized by edema,
heavy proteinuria (>1 g/m2
daily; >40 mg/m2/hr) and hypoalbuminemia (serum albumin
<3 g/dL), is among the most common kidney diseases in childhood.
The condition has an annual incidence ranging from 1.2 to 16.9
per 100,000 children [1,2]. While nephrotic syndrome is usually
primary or idiopathic, evaluation might reveal an underlying
systemic illness in 5-10% of patients. Kidney biopsy reveals
minimal change disease in ~80% patients, and focal segmental
glomerulosclerosis (FSGS) and mesangioproliferative
glomerulonephritis (GN) in 7-8% each. Therapy with prednisolone
results in complete remission of proteinuria in 85-90% patients,
termed steroid sensitive nephrotic syndrome (SSNS). While the
outcome in patients with SSNS is satisfactory, approximately 50%
show frequent relapses or steroid dependence, and 3-10% show
late steroid resistance [3-5].
OBJECTIVE
Guidelines on management of SSNS, by the
Indian Society of Pediatric Nephrology, were first published in
2001 [6] and updated in 2008 [7]. With increasing availability
of evidence on various therapies, these guidelines have been
revised. Guidance is based on the strength and quality of
evidence using the GRADE model proposed by the American Academy
of Pediatrics [8]. Ungraded statements (indicated by X) are like
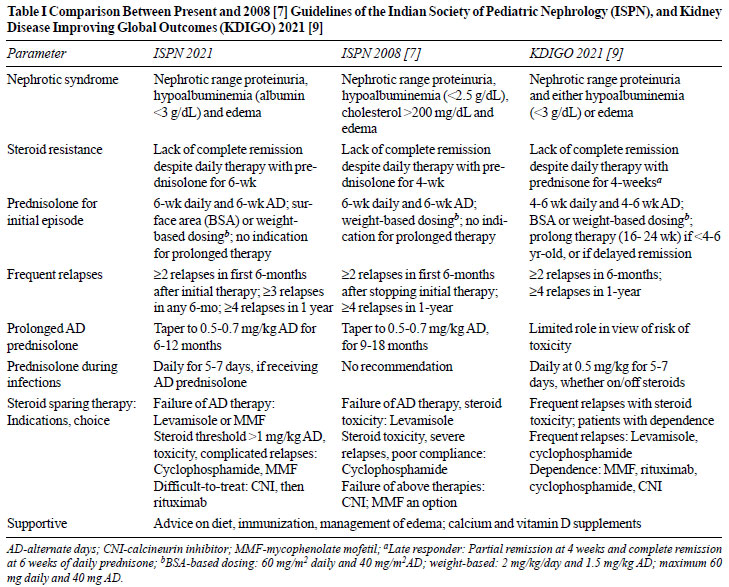
practice points, not supported by sufficient evidence. Table
I highlights key changes in present guidelines compared to
2008 [7] and those recently proposed by the Kidney Disease
Improving Global Outcomes [9].
PROCESS
Workgroups were constituted to address key
issues, including: (i) Evaluation at baseline and follow
up, role of biopsy, genetic testing, and differential diagnosis;
(ii) Management of the initial episode and subsequent
relapses; (iii) Management of frequent relapses; and
(iv) Supportive care and outcomes. Separate workgroups have
addressed guidelines on the definition and management of steroid
resistant nephrotic syndrome [10]. The workgroups identified
gaps in knowledge, formulated questions and developed consensus
statements prior to the meeting in New Delhi on 5 April 2019,
when the evidence was discussed through alternating breakout and
plenary sessions. Research studies were rated from A to D using
standard criteria, and each consensus statement was assigned one
of two levels of recommendation, based on assessment of relative
benefit versus harm, and relevance in context of
availability and cost, and the feasibility of monitoring (Supp.
Table I) [11]. Draft guidelines were again discussed
in Pune on 21 December 2019. The final manuscript was circulated
to all participants for approval.
DEFINITIONS
Criteria for defining the course of nephrotic
syndrome are shown in Box I [12-14]. For
purpose of this guidelines, unless stated, the term ‘frequent
relapses’ includes patients with ‘steroid dependence’, and
prednisolone and prednisone are used interchangeably. The
management of initial and late resistance, defined as lack of
remission following 6-weeks’ prednisolone therapy (Box
I) is discussed separately [10].
Patients with frequent relapsing and steroid
resistant nephrotic syndrome are at high risk of complications,
due to the illness and toxicity of medications. We advise that
these patients, and those younger than one year, be managed by
pediatric nephrologists.
Guideline 1: Evaluation
1.1 In a patient presenting with recent
onset of edema, we recommend the following investigations to
confirm the diagnosis of nephrotic syndrome: (i)
urinalysis; and (ii) blood levels of urea,
creatinine, albumin and total cholesterol (Box II).
(X)
1.2 We suggest additional evaluation in
selected patients (Box II). (X)
1.3 We recommend that parents be taught
to maintain a record of proteinuria (by dipstick or
boiling), infections and medications received. (X)
Rationale
Children with the first episode of nephrotic
syndrome require evaluation to confirm the diagnosis and screen
for an underlying cause and complications. Family history of
nephrotic syndrome, asthma and allergies, and renal diseases are
asked for. Features including fever, abdominal pain, rash,
arthralgia, oliguria, hematuria and history of drugs or
infections suggest an underlying cause, e.g., systemic lupus
erythematosus and IgA vasculitis. Height, weight and blood
pressure should be recorded; weight monitoring helps in
assessment for edema.
Investigations advised at the initial episode
are listed in Box II. The diagnosis is based on
presence of nephrotic range proteinuria, hypoalbuminemia and
edema. Majority of patients show total cholesterol levels
exceeding 200 mg/dL. Nephrotic range proteinuria is present if
in an early morning urine sample protein is 3-4+ (dipstick/
boiling test), spot protein to creatinine ratio is >2 mg/mg, or
the protein excretion is >40 mg/m 2
per hr. Precise estimation of 24-hr protein excretion is
cumbersome, and is seldom necessary. Urine microscopy is normal,
except for hyaline or granular casts; occasional microscopic
hematuria is not uncommon. Persistent microscopic hematuria or
red cell casts suggests disease other than minimal change
nephrotic syndrome, like infection related GN, C3
glomerulopathy, systemic lupus or vasculitis [1]. Additional
investigations are required for their diagnosis. Since patients
with nephrotic syndrome do not have increased prevalence of
urinary tract infections, routine urine cultures are not
necessary.
With an estimated prevalence of
bacteriologically positive pulmonary tuberculosis of 296 per
100,000 population in India, the risk of latent tuberculosis
infection in childhood is high [15,16]. Tuberculin test is
suggested prior to the first course of steroid treatment,
especially with history of contact [16]. Chest radiography is
done in patients with positive tuberculin test; those with
features of tuberculosis require appropriate therapy. Patients
with positive tuberculin reaction, but no radiological or
bacteriological evidence of tuberculosis, should receive
isoniazid prophylaxis for 6-months [16]. The prevalence of
hepatitis B in non-tribal Indian populations is low (2.4%; 95%
CI, 2.2-2.7%) [17], and routine screening is not required.
Genome wide association studies have
identified variants in multiple MHC class II molecules as risk
factors for SSNS [18]. The diagnostic and prognostic utility of
various biomarkers of minimal change disease is limited [19].
There is, currently, no role for biomarkers or genetic studies
in these patients.
Subsequent Evaluation
Parents are instructed to monitor the child’s
urine at home, using dipstick or boiling test, and are explained
the features of a relapse. During remission, they are advised to
screen for proteinuria 2-3 times a week; the child is also
examined every day during infections, or if edema is present.
Frequent assessment of biochemistry is not necessary. Evaluation
of patients during relapses also includes screening for
complications (Box II).
Guideline 2: Kidney biopsy
2.1 We recommend kidney biopsy in
nephrotic syndrome, in the presence of: (i)
persistent microscopic hematuria, gross hematuria, or acute
kidney injury not attributed to hypovolemia; (ii)
systemic features: fever, rash, arthralgia, low complement
C3; (iii) initial or late corticosteroid resistance;
and (iv) prolonged (>30-36 months) therapy with
calcineurin inhibitors (CNI), or reduced kidney function
during their use. (1B)
2.2 We suggest performing kidney biopsy
prior to initiating therapy with CNI. (X)
2.3 We recommend light microscopy and
immuno-fluorescence examination on all kidney biopsies.
Electron microscopy is required in patients with gross or
persistent microscopic hematuria, low C3 and suspected
disorders of glomerular basement membrane. (X)
Rationale
Clinicopathological studies show that kidney
biopsy is not routinely required in children with idiopathic
nephrotic syndrome prior to therapy with corticosteroids
[20-22]. Remission of proteinuria following steroid therapy is
the most important predictor of long-term outcome [3,23]. The
chief indication of kidney biopsy is in patients who fail to
show complete remission of proteinuria despite 6-weeks daily
therapy with prednisolone (steroid resistant illness) [10,24]. A
biopsy is indicated in patients with gross hematuria or
persistent microscopic hematuria at the onset (> 5 red cells per
high power field on 3 or more occasions, in urine centrifuged at
400 g for 4-5 minutes); or extrarenal features of a systemic
disease [20-23,25].
An age of onset of more than 12-years is
often cited as an indication for performing a kidney biopsy.
Review of literature in adolescent onset nephrotic syndrome
suggests that a combination of features, including persistent
microscopic hematuria, low C3 and steroid resistance, detects
all patients with membranous nephropathy or proliferative GN
[20-22,26,27]. This might obviate the need for a kidney biopsy
in adolescents presenting with typical nephrotic syndrome that
is steroid sensitive. Since infants (<12-months-old), including
those with congenital nephrotic syndrome, are likely to show
histological features other than minimal change disease or an
underlying genetic change, we advise next-generation sequencing
in these patients [10]. Patients with onset of idiopathic
nephrotic syndrome beyond infancy should receive therapy with
prednisolone, and are advised to undergo kidney biopsy if they
show steroid resistance.
The large majority of patients with SSNS show
minimal change disease, and less commonly, FSGS or
mesangioproliferative GN [20-22,28]. More than 90% children with
minimal change disease, 50% with mesangioproliferative GN, and
30% with FSGS have steroid sensitive disease. Patients with
frequent relapses do not require a biopsy before initiating
therapy with steroid-sparing agents like levamisole,
cyclo-phosphamide, mycophenolate mofetil (MMF) or rituximab
[29]. The exception is prior to the use of CNI.
While there is limited guidance to support
kidney biopsy in patients with SSNS prior to the therapy with
CNI [9,30], information on the extent of tubular atrophy and
interstitial fibrosis is useful when planning therapy. Therapy
with CNI might result in acute nephrotoxicity, manifested as
acute tubular injury and isometric tubular epithelial
vacuolization [31,32]. Chronic nephrotoxicity, characterized by
striped tubulointerstitial fibrosis has been reported in 25-43%
biopsies following therapy (for 2.5-3.5 years) with cyclosporin
or tacrolimus [33-35]. While a recent report found low risk of
nephrotoxicity despite prolonged use of tacrolimus [36], most
reports suggest similar risk with both medications [34,37]. We
therefore suggest considering kidney biopsy before initiating
therapy with CNI, particularly in patients with prolonged
disease and unclear course, and to inform the clinician
regarding baseline histological changes and allow appropriate
counseling. In view of long-term risks of nephrotoxicity, kidney
biopsy should be performed following prolonged therapy with CNI,
or if the therapy is associated with decline in eGFR that
persists despite reduction in CNI dose [9,38].
An adequate biopsy specimen should preferably
include the corticomedullary junction and approximately 20
glomeruli to exclude the diagnosis of FSGS [39]. Apart from
renal histology, the biopsy provides information on extent and
morphology of glomerulosclerosis and associated
tubulointerstitial changes. The diagnosis of IgA nephropathy, C3
glomerulopathy and early membranous nephropathy is suggested by
immunofluorescence studies. While kidney biopsies from all
patients with nephrotic syndrome should be examined by electron
microscopy, the facility is often not available. Ultrastructural
examination helps to confirm the diagnosis of minimal change
disease (effacement of podocyte foot processes; no electron
dense deposits), differentiate primary from secondary FSGS
(diffuse versus focal foot process effacement), categorize
membranous nephropathy and C3 glomerulopathy, and identify
disorders of glomerular basement membrane [40].
Guideline 3: Therapy for the first episode of
nephrotic syndrome
We recommend that therapy for the initial
episode should comprise of prednisolone at a dose of 60 mg/m 2/day
(2 mg/kg/day, maximum 60 mg in 1-2 divided doses) for 6 weeks,
followed by 40 mg/m2 (1.5 mg/kg, maximum 40 mg as single morning dose)
on alternate days for the next 6 weeks, and then discontinued.
(1A)
Rationale
In 1981, the International Study of Kidney
Disease in Children (ISKDC) proposed that the first episode of
nephrotic syndrome be treated with daily prednisone for 4-weeks,
followed by intermittent therapy for the next 4-weeks, and then
discontinued [41]. Later, a randomized controlled trial (RCT) by
the Arbeitsgemeinschaft für Padiatrische Nephrologie showed that
therapy with prednisolone for 6-weeks daily and 6-weeks
alternate-day was better in terms of reduced incidence of
relapses over the next 12-24 months [42]. In efforts to define
optimal therapy for the initial episode, several RCTs have
investigated the duration and dose of prednisolone, based on
which, a meta-analysis, in 2007, concluded that prolonging
therapy for 6-months was associated with reduced risk of
relapses and of frequent relapses (relative risk, RR 0.55; 95%
CI 0.39-0.80) [43]. However, most studies included in this
analysis had methodological flaws, resulting in a high risk of
bias.
Four large multicenter RCTs published in the
last 7 years have challenged the previous results
(Supp.
Table II). These studies, representing outcomes in
over 800 patients across Netherlands, UK, Japan and India, show
that extending initial therapy beyond 8-12 weeks does not
influence either the time to first relapse or the risk of
frequent relapses at 1-2 years’ follow up. These studies had low
risk of bias; three were placebo-controlled. A meta-analysis
that included three of these studies, showed that the risk of
frequent relapses at 1-2 years’ follow-up was lower for 3-months
or longer versus 2-months therapy (RR 0.68; 95% CI 0.47-1.0),
but not for 5-months or longer versus 3-months therapy (RR 0.78;
95% CI 0.50-1.22) [44]. Subgroup analysis, limited to studies at
low risk of bias, indicated similar risk for frequent relapses
in patients treated for 2-3 months versus 3-6 months. These
findings are confirmed with inclusion of the PREDNOS study (Supp.
Fig. 1) [45]. While post-hoc analyses in two
studies suggest a trend for benefit with prolonged therapy in
young children, this finding requires confirmation [45,46].
Based on pharmacokinetics and variations by
age, prednisolone is preferably dosed by body surface area in
children [47]. However, estimation of body surface area involves
complex formulae with variable results [48]. Calculation using
body weight is convenient, but results in relative underdosing,
particularly in young children [47,49]. Underdosing, using
weight-based calculations, was associated with increased risk of
frequent relapses in some [50,51], but not in all studies
[52,53]. Experts therefore prefer to administer prednisolone
based on body surface area for young children [47].
Daily prednisolone is administered in single
or divided-doses, with similar time to remission [54]. There is
no evidence to support therapy with preparations other than
prednisone or its active metabolite, prednisolone [55]. Use of
deflazacort, betamethasone, dexamethasone or methylprednisolone
is not advised. Prednisolone is best given following food;
therapy with antacids, ranitidine or proton pump inhibitors is
not routinely required.
Guideline 4: Therapy of relapses
We recommend that relapses be treated with
prednisolone at 60 mg/m 2/day
(2 mg/kg/day; maximum 60 mg) in single or divided-doses until
remission (protein trace/nil for 3 consecutive days), followed
by 40 mg/m2 (1.5
mg/kg, maximum 40 mg) on alternate days for 4-weeks. (1C)
Rationale
Almost one-half of the relapses are
precipitated by minor infections, usually of the upper
respiratory tract. Treatment of infection may rarely induce
remission, avoiding the need for corticosteroid therapy. A
relapse has conventionally, albeit empirically, been treated as
outlined above, but guidelines vary in the duration of therapy.
Remission is achieved by 7-10 days, and daily therapy is seldom
necessary beyond 2 weeks. In case of persistent proteinuria,
daily therapy with prednisolone may be extended, to maximum of
6-weeks. Lack of remission despite treatment with 6-weeks’ daily
prednisolone indicates late steroid resistance that requires
specific evaluation and management [10].
Dose based on body surface area and weight is
associated with similar time to remission and frequency of
subsequent relapses [52,53]. Retrospective studies and small
RCTs suggest that reduced dose or abbreviated duration of
therapy with prednisolone is effective in inducing and
maintaining remission (Supp. Table III). Well-powered
studies are required to evaluate the optimal dose and duration
of prednisolone for relapses.
Guideline 5: Management of frequent relapses
and steroid dependence
Definition
Frequent relapses are defined by the ISKDC as
occurrence of two or more relapses in the first 6-months after
initial response, or four or more relapses in a year [3]. These
patients are at risk of morbidity associated with multiple
relapses and corticosteroid toxicity. The term has been used for
over 40-yr, with minor modifications. Additionally, we propose
that patients with three or more relapses in any 6-months be
also classified as frequent relapsers (Box I).
Steroid dependence, as previously defined, includes patients
with two consecutive relapses, while receiving or within 2-weeks
of discontinuing prednisolone [3,6].
The occurrence of two or more relapses in the
first 6-months is usually associated with high frequency of
relapses in the subsequent 12-24 months [3]. Patients
experiencing 4 relapses annually receive ~165-200 mg/kg (4.6-5.6
g/m 2) prednisolone,
corresponding to 0.45-0.55 mg/kg (12.5-15.5 mg/m2)
daily. As 12-weeks’ prednisolone therapy for the initial episode
(~115 mg/kg; ~3.4 g/m2)
might be associated with adverse effects [55,56], the risk of
steroid toxicity in patients with 3 relapses in any 6-months or
4 relapses annually is considerable [57].
Two additional situations might suggest the
need for steroid-sparing therapy. The first is a patient with
significant steroid toxicity (Box I) and fewer
relapses (3 relapses/year; 2 relapses in 6-months). The second
is the occurrence of two relapses in 6-months during long-term
therapy with corticosteroids or steroid-sparing agents. In both
instances, it is rational to manage the patients as frequent
relapsers, even if they do not satisfy standard definitions.
While stable remission (sustained remission or infrequent
relapses i.e., upto one relapse in 6-months) during therapy with
steroid-sparing agents is acceptable, the definition of failure
of therapy depends on the medication, interval between relapses
and need for concomitant corticosteroids.
5.1 Choice of therapy
We recommend that the choice of
immunosuppressive strategy for patients with frequent relapses
be based on considerations of its efficacy and adverse effects,
patient age, steroid threshold, severity of relapses and
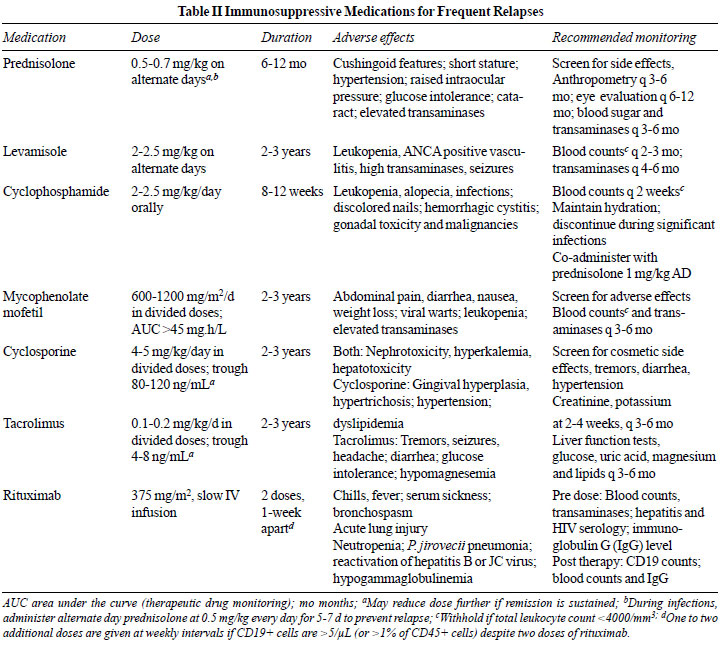
features of steroid toxicity (Fig. 1). (X)
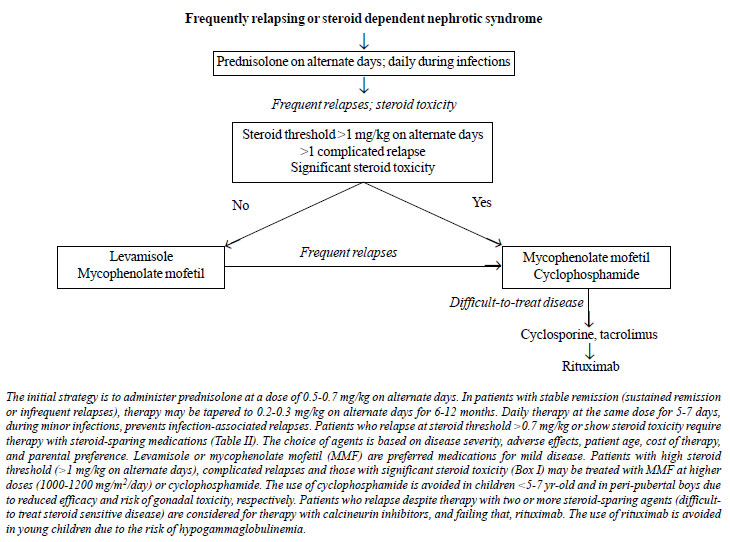 |
|
Fig. 1. Management of
frequently relapsing or steroid dependent nephrotic
syndrome.
|
Rationale
In patients with frequent relapses,
guidelines recommend that corticosteroid therapy for the relapse
be prolonged and tapered over 3 months or longer [9,30,58]. The
dose at which relapses occur (steroid threshold) is a marker of
disease severity. Prolonged therapy with alternate-day
prednisolone might maintain remission in patients with low
threshold relapses (<0.7 mg/kg on alternate days).
Steroid-sparing interventions are necessary
in patients who continue to relapse frequently or show evidence
of steroid toxicity while on alternate-day prednisolone (Fig.
1). There is limited data on relative efficacy of
various steroid-sparing agents, and the choice of
immunosuppressive strategy is guided by its efficacy, safety,
cost and availability, patient age, disease severity, and
parental preference (Table II). Potent medications
are preferred in patients with high threshold (>1 mg/kg on
alternate day) relapses, relapses associated with
life-threatening complications, or with significant steroid
toxicity (Box I and Table II). The presence
of stable remission (up to one relapse in 6 months) during such
therapy is acceptable, and except in severe steroid dependence,
prednisolone is tapered and discontinued over few months.
Therapy may be modified in patients with frequent relapses or
significant adverse effects.
 |
A proportion of patients with SSNS show
disease characterized by multiple relapses despite therapy with
steroid-sparing agents, and/or medication-associated toxicity.
We propose defining difficult-to-treat nephrotic syndrome as
patients with: (i) frequent relapses or infrequent
relapses with significant steroid toxicity; and (ii)
failure of 2 or more steroid sparing agents: levamisole,
cyclophosphamide or MMF. These patients might merit therapy with
agents such as CNI and rituximab.
While the approach to management indicated in
Fig. 1 suffices in most instances, individual
situations may require different preference. Patients diagnosed
either with steroid dependence soon after initial therapy, or
with significant steroid toxicity at diagnosis of frequent
relapses may be considered directly for steroid sparing
therapies. Therapy with oral cyclophosphamide is avoided in
young patients and in pubertal or post-pubertal boys. Therapy
with CNI may be preferred to MMF in very young patients with
significant steroid toxicity, even though the definition of
difficult-to-treat SSNS is not met.
5.2 Long-term corticosteroids
• In patients with frequent relapses, we
suggest tapering prednisolone to a dose of 0.5-0.7 mg/kg on
alternate days, for 6-12 months. (2B)
• In patients receiving long term
alternate-day prednisolone, we recommend administering the
same dose daily for 5-7 days during fever or respiratory
tract infection. (1B)
Rationale
Therapy with alternate-day prednisolone is
the initial strategy for managing patients with frequent
relapses [6,58]. Alternate-day prednisolone, often used as the
control limb in RCTs, showed satisfactory response in 43-82.5%
patients (Supp. Table IV). A balance of benefit
over harm is lacking, and there are risks of corticosteroid
toxicity. Therefore, in patients in remission at prednisolone
dose of 0.5-0.7 mg/kg for a few months, the medication may be
tapered to ~0.2-0.3 mg/kg on alternate days. The duration of
therapy is at physician discretion, based on its efficacy and
assessment of toxicity through monitoring of weight, height,
blood pressure, ocular toxicity and hyperglycemia (Table
II).
Daily prednisolone during infections
More than one-half of relapses in SSNS occur
following upper respiratory tract infections. Evidence from
three studies (Supp. Table V) indicates that,
beginning with the onset of infection, switching therapy from
alternate-day to daily administration of prednisolone for 5-7
days prevents the occurrence of relapses. One cross-over trial
also supports the use of low-dose daily prednisolone in
preventing infection-associated relapses in patients off
corticosteroids [59]. Results of the PREDNOS2 trial will clarify
the role of these strategies in preventing infection-associated
relapses (ISRCTN10900733).
Daily prednisolone in low-dose
Data from an open-label RCT [60] and a case
series [61] suggests that low-dose (0.2-0.3 mg/kg) daily
prednisolone is associated with fewer relapses than twice the
dose (0.5-0.7 mg/kg) on alternate days. The strategy led
to lower steroid requirement and was not associated with
toxicity [60]. These findings require confirmation in studies
with longer follow-up that are powered to examine adverse
effects, including suppression of the
hypothalamo-pituitary-adrenal axis [62].
5.3 Non-corticosteroid therapies
• We recommend use of a steroid-sparing
agent in patients failing therapy with alternate-day
prednisolone, steroid toxicity or complicated relapses (Fig.
1). (1B)
• In patients failing alternate-day
prednisolone, we recommend therapy with either levamisole or
MMF for 12-24 months. (1B)
• We recommend MMF or cyclophosphamide in
patients with significant steroid toxicity, high steroid
threshold, complicated relapses, of failure of therapy with
levamisole. (1C)
Rationale
Levamisole:
Levamisole has been used for
almost 4-decades, mainly in Asia and Europe, as a
steroid-sparing agent for frequent relapsing nephrotic syndrome
[63]. A meta-analysis (8 studies, 462 patients;
Supp. Table
VI), suggests 35% reduction in the risk of relapses
following 6-12 months’ therapy with levamisole (RR 0.65; 95% CI
0.48-0.88) [64]. The medication is more useful in patients with
frequent relapses than in steroid dependence [65]. Comparative
studies indicate that the risk of relapse in patients receiving
levamisole is similar to cyclophosphamide (2 studies, 97
children; RR 2.14; 95% CI 0.22-20.95), or MMF (one study, 149
patients; RR 1.11; 95% CI 0.86-1.43) [64]. Given the efficacy
and safety, the agent is being examined in two RCTs when
administered at onset of the disease (LEARNS, EudraCT
2017-001025-41; NEPHROVIR3, NCT02818738).
Levamisole is given at the dose of 2-2.5
mg/kg on alternate days (Table II). While few
retrospective studies report its efficacy when administered
daily (Supp. Table VII), the safety of this
strategy should be examined in controlled studies with close
monitoring for adverse effects, including neutropenia, raised
transaminases, anti-neutrophil cytoplasmic antibodies and/or
small vessel vasculitis [63,66,67].
Mycophenolate mofetil (MMF):
The use of MMF in frequently relapsing
nephrotic syndrome is recent [68]. A review of 7 prospective and
6 retrospective series (508 patients) showed that therapy with
MMF for 6-19 months lowered relapse rates, and reduced
requirement of prednisolone and/or CNI (Supp. Table VIII)
[68]. While placebo-controlled, blinded RCTs are lacking, MMF
was found to be comparable to levamisole but inferior to
cyclosporine in maintaining satisfactory remission or reducing
the frequency of relapses in 3 open-label RCTs (Supp. Table
IX) [64]. Likewise, MMF had efficacy similar or inferior to
tacrolimus in a non-randomized comparison (Supp. Table IX).
MMF is perhaps more efficacious in young children [69], and more
effective than levamisole in patients with steroid dependence
[70].
Therapy with MMF is given in two divided
doses, 600 to 1200 mg/m2
(20-30 mg/kg) daily [68]. Dose-related adverse effects include
leukopenia, abdominal pain and diarrhea. Data from one RCT
suggests that patients with higher blood levels of MMF
(determined by area under the curve, AUC) show efficacy similar
to cyclosporine [71]. Others emphasize the need to achieve
mycophenolic acid AUC levels exceeding 45-60
mg*h/mL
[72-74] or trough levels >2-3
mg/mL
[75-78]. While pharmacokinetics of MMF is variable, adequate
levels are achieved with high doses [76-78]. In the absence of
facilities for therapeutic drug monitoring, we propose
initiating therapy at the lower end of dose range and escalating
as tolerated, to 1000-1200 mg/m2,
if the patient continues to relapse.
Cyclophosphamide:
Oral cyclophosphamide, at 2-2.5
mg/kg daily for 8-12 weeks, is the most commonly used
steroid-sparing agent in SSNS. Its use finds basis in evidence
of efficacy and overall safety, as summarized in a systematic
review (38 prospective and retrospective studies, 1504 patients)
of patients administered cyclo-phosphamide or chlorambucil [79].
A recent meta-analysis shows reduced risk of relapse at 6-12
months (6 studies, 202 patients; RR 0.44; 95% CI 0.32-0.60) and
12-24 months (4 studies, 59 patients; RR 0.20; 95% CI 0.09-0.46)
following therapy with alkylating agents [64]. In comparative
studies, the risk of relapse at 12-24 months following
cyclophosphamide therapy was similar to levamisole (1 study, 40
patients; RR 1.12; 95% CI 0.86-1.16), but lower than
cyclosporine (2 studies, 95 patients; RR 0.51; 95% CI 0.35-0.74)
[64]. A Bayesian network analysis (7 reports, 391 patients)
showed lowest relapse rates with cyclophosphamide, compared to
other medications [80]. Cyclophosphamide is more effective in
patients with frequent relapses than in steroid dependence, and
in patients older than 5-7 years (Supp. Table X).
Therapy with cyclophosphamide is initiated
during remission. Prednisolone is given at a dose of ~1 mg/kg on
alternate days during therapy with cyclophosphamide; the
medication may subsequently be stopped after 1-2 months.
Leukopenia is the chief adverse effect, reported in one-third of
patients; other concerns are alopecia and the risk of infections
(Table II). Leukocyte count is monitored every 2
weeks, and therapy withheld if the count falls below 4000/mm3.
Increased fluid intake and frequent voiding prevents hemorrhagic
cystitis which, along with nausea and vomiting, is common with
intravenous (IV) dosing. The risk of gonadal toxicity is
proportionate to the cumulative dose, and appears to be high in
pubertal and post-pubertal boys (Tanner stage 2 or more), and
lower in girls [30,79,81]. Therapy with chlorambucil is
associated with risk of seizures, and is not recommended.
Given concerns of gonadal toxicity and
malignancy, therapy with cyclophosphamide is usually
administered after failure of levamisole or MMF, and is limited
to one 12-weeks’ course (cumulative ~168 mg/kg). Occasionally,
cyclophosphamide may be the preferred initial steroid-sparing
therapy in patients older than 7-yr, particularly in presence of
significant steroid toxicity and/or complicated relapses.
Limited evidence indicates that cyclophosphamide (500 mg/m2
monthly IV pulse; 6-doses) is as effective as 12-weeks’ oral
therapy [64], and may be considered in patients with likely
non-compliance to oral therapy.
5.4 Difficult-to-treat steroid sensitive
nephrotic syndrome
• We recommend therapy with CNI, either
cyclosporine or tacrolimus, in patients with
difficult-to-treat SSNS. (1B)
• We recommend therapy with rituximab in
patients who have either failed CNI or have received these
agents for a prolonged duration. (1C)
• We suggest that therapy with rituximab
be administered during disease remission after ruling out
acute and chronic infections, and should target B cell
depletion. (2B)
Rationale
Calcineurin inhibitors:
Observational studies indicate that CNI (cyclosporine 4-6
mg/kg/day, tacrolimus 0.1-0.2 mg/kg/day, in two divided doses)
maintain remission and enable steroid-sparing in 60–90% patients
with frequent relapses or steroid dependence who have failed
treatment with alkylating agents [82-84]. These agents have not
been compared to placebo or to each other in controlled studies
for SSNS. While one RCT each found that cyclosporine was
associated with reduced risk of relapse as compared to
prednisolone (104 children; RR 0.33; 95% CI 0.13-0.83) or MMF
(see above), patients relapsed when the therapy was discontinued
[64]. In view of the efficacy and significant steroid-sparing,
CNI are preferred for patients with high threshold relapses or
significant corticosteroid toxicity. While therapy with CNI is
usually restricted to patients with difficult-to-treat SSNS (Box
I), these agents may be considered before MMF or
cyclophosphamide in young children with severe steroid
dependence and/or significant steroid toxicity. The choice of
the medication should follow discussion with parents about
potential toxicities and the need for monitoring.
Chief adverse effects of CNI include acute
and chronic nephrotoxicity (with both agents), hirsutism, gum
hypertrophy, hypertension and hyperlipidemia (with
cyclosporine), and hyperglycemia or seizures (with tacrolimus)
[82,83]. While tacrolimus is preferred to cyclosporine due to
lack of cosmetic effects, only the latter is available as an
oral suspension for young children. Therapy should be
administered for at least 12-months, with monitoring of drug
levels (Table II). Lower target trough levels and
once-daily dosing is acceptable during sustained remission
[85.86]. The role of protocol biopsies, before initiating
therapy with CNI and following their prolonged use, is discussed
in Guideline 2.
Rituximab: B cell depletion has emerged
as an effective strategy for sustaining remission in patients
with steroid- and/or CNI-dependent nephrotic syndrome. Therapy
with rituximab (375 mg/m2
IV once a week for 1-4 doses) in 13 prospective and
retrospective series (n=159) led to sustained remission in
25-71% patients, postponement of relapse by (median) 5-11
months, and withdrawal of other therapies [87]. A systematic
review confirmed similar efficacy in 86 adults administered
rituximab for frequent relapses [88]. In non-randomized
comparisons, the efficacy of rituximab was superior to
cyclophosphamide (2 studies, 148 patients) and comparable to
tacrolimus (1 study, 23 patients) (Supp. Table XI). In a
prospective study, therapy with 2-3 doses of rituximab in 101
patients was associated with over two-third reduction in
relapses, postponement of relapse by median 16-months and
reduced steroid requirement [89].
Data from 7 RCTs in patients with frequent
relapses and steroid/CNI dependence indicates superior efficacy
of rituximab as compared to placebo (2 studies, 71 patients), or
no additional therapy (2 studies, 91 patients); the efficacy was
similar or superior to CNI in one study each (174 patients) (Supp.
Table XI). A Cochrane meta-analysis concluded that
therapy with rituximab, in combination with CNI and
prednisolone, versus the latter alone, reduced the risk
of relapse at 6 months (5 studies, 269 patients; RR 0.23, 95% CI
0.12-0.43) and 12 months (3 studies, 198 patients; RR 0.63, 95%
CI 0.42-0.93) [64].
Experts advise administering rituximab at a
dose of 375 mg/m2
IV, using B cell depletion (CD19+ cells <1% of CD45+ cells, or
<5 cells/µL) as a marker for adequacy of dosing. While B cell
depletion is usual after even one dose [87], a maximum of 4
infusions have been given. Since administration of rituximab
during relapse is associated with its urinary excretion and
reduced half-life, therapy is preferred during remission [90]. B
cell recovery usually occurs by 6-9 months, and is associated
with risk of relapses [87,88,90]. Studies comparing response to
rituximab in relation to the number of doses and use of
maintenance immunosuppression are summarized in
Supp. Table
XII. An international cohort on 511 patients with
frequent relapses or steroid dependence showed that relapse-free
survival was significantly shorter for patients given a single
dose of rituximab (8.5 months) compared to those given two (12.7
months) or more doses (14.3 months) [91]. Additional
immunosuppression was useful in sustaining remission following
therapy with a single dose of rituximab. In patients with
difficult-to-treat SSNS with satisfactory response to rituximab,
repeated doses of the medication, following relapses or
repopulation of B cells, is suggested as a strategy to sustain
remission (Supp. Table XII). Given the concerns
discussed below, the optimal strategy is still not clear.
Systematic reviews show that therapy with
rituximab is associated with infusion reactions (4 studies, 252
children; RR 5.8, 95% CI 1.3-25.3) [64], delayed adverse events
and infections [87,88]. A German registry of autoimmune diseases
(370 patients) reported serious infections in 5.3 cases per 100
patient-years [92]. Patients with lymphoma treated with
rituximab show reactivation of hepatitis B virus infection in 9%
(95% CI 5%-15%) patients [93]. In contrast to the reports of
normal IgG in adult patients receiving multiple doses of
rituximab (Supp. Table XII), hypogammaglobulinemia
is not uncommon in children with nephrotic syndrome and
autoimmune diseases. The risk of hypogammaglo-bulinemia
correlates inversely with age, and positively with the number of
rituximab doses [94-96].
We recommend that rituximab be used in
patients with difficult-to-treat disease, under the supervision
of a pediatric nephrologist. Its use should be avoided in young
children (<5-7 yr old), and restricted to patients failing other
steroid-sparing agents. Active acute infections and chronic
viral infections should be ruled out before therapy. We
recommend administering two doses of rituximab during disease
remission, at 375 mg/m2
one-week apart, followed by confirmation of B cell depletion,
2-7 days after the second dose. Vigilance for infections and
monitoring for leukopenia and hypogamma-globulinemia is
essential during follow up. Further doses of rituximab should be
avoided in patients with severe infusion-related adverse events,
severe infections or with hypogammaglobulinemia. Prophylactic
antibiotics are not routinely recommended. We suggest
administering cotrimoxazole (150 mg/m2
or 5 mg/kg of trimethoprim on alternate days) in patients
receiving additional immuno-suppression, such as those receiving
maintenance treatment with CNI or MMF following therapy with
rituximab.
SUPPORTIVE CARE
Patients with nephrotic syndrome are at risk
of complications of the disease, and side effects of its
medications. Principles of management of hyper- tension,
thromboembolism, growth retardation, obesity, dyslipidemia, and
hypothyroidism are discussed in the guidelines on steroid
resistant nephrotic syndrome [10]. We emphasize that patients
who have received oral steroids for more than 2-weeks within the
past one-year, should receive additional corticosteroids during
conditions associated with physiological stress like systemic
infections, inadequate oral intake, lethargy, dehydration,
invasive or dental surgery, trauma and large burns [10].
Conditions such as uncomplicated viral infections, acute otitis
media and fever following immunization do not require stress
dosing with steroids.
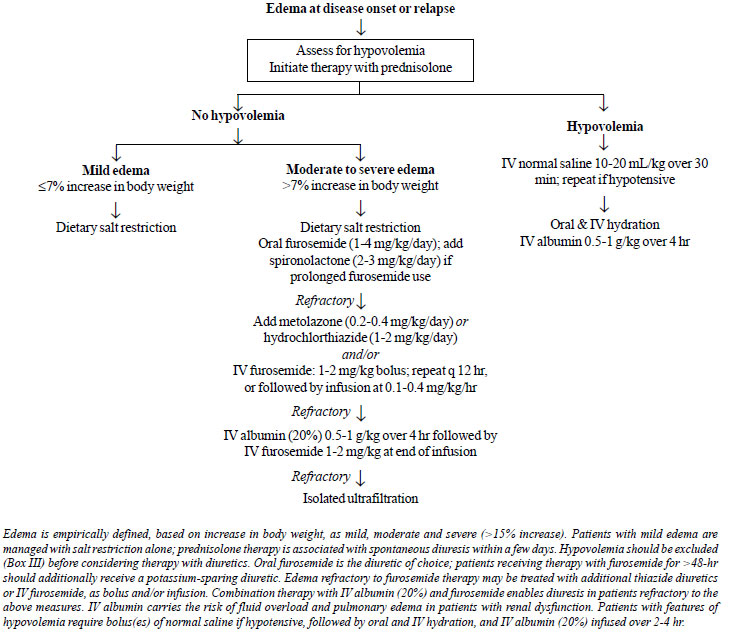
Guideline 6: Management of Hypovolemia and
Edema
Edema, a cardinal feature of nephrotic
syndrome, often requires specific therapy. We propose that edema
be empirically classified based on appearance and percentage
weight gain from baseline, as mild ( £7%
increase), moderate (8-15%) and severe (>15% increase) [97]. If
urine protein is monitored regularly, the occurrence of more
than mild edema is unusual. Patients with severe edema have
marked hypoalbuminemia (serum albumin <1.5 g/dL), along with
ascites and anasarca that interferes with daily activities
[97,98]. Intravascular volume depletion is common in patients
with moderate or severe edema [99,100], and should be assessed
before instituting therapy with diuretics.
6.1 Hypovolemia
• We recommend that patients with
moderate to severe edema be assessed for intravascular
volume status before initiating therapy with diuretics (Fig.
2). (X)
• We recommend the use of normal saline
and IV albumin in patients with disease relapse and
hypovolemia. (1C)
Rationale
A combination of clinical and biochemical
features helps estimate intravascular volume (Box III,
Fig. 2) [97,101]. Patients with hypovolemia often
have abdominal pain, nausea, vomiting, dizziness and lethargy.
Examination shows tachycardia, pallor, cold peripheries, delayed
capillary refill and postural hypotension, and rarely shock
[97,101,102]. On the other hand, patients with hypervolemia have
refractory anasarca, hypertension and dyspnea [99,100]. Two
urinary indices may help assess intravascular volume: fractional
excretion of sodium (FENa) and potassium index [103,104]. While
both underfill and overfill states are associated with sodium
retention [105-107], FENa <0.5% and potassium index >0.6
indicate high aldosterone activity, characteristic of
hypovolemia [104,105,108]. The indices are not reliable with
recent diuretic therapy and while receiving IV fluids. Other
parameters of volume status include changes in hematocrit, urea
to creatinine ratio [105], inferior vena cava diameter and
collapsibility, and bioimpedance analysis [97,99-101,109,110].
 |
Hypovolemia may occur at disease onset or
relapse, particularly in a setting of diarrhea, vomiting or
unsupervised diuretic therapy. Therapy with diuretics should be
discontinued. Hypotensive patients should receive 1-2 boluses of
isotonic saline (10-20 ml/kg infused over 20-30 minutes) and/or
5% albumin (10–15 ml/kg over 30-60 minutes) (Fig. 2).
Subsequently, patients are managed with IV and oral hydration,
and IV albumin (20%; 0.5–1 g/kg over 3-4 hr) [97,99,101].
 |
|
Fig. 2. Management of edema in
nephrotic syndrome.
|
6.2 Edema
• We recommend oral furosemide as first
line therapy for patients with moderate edema without
hypovolemia (Fig. 2). (1C)
• We suggest that patients with
furosemide-refractory edema be managed as follows: (i)
combination of loop diuretics with thiazide; (ii)
co-administration of human albumin with IV furosemide. (X)
Rationale
Patients with mild edema do not require
diuretic therapy. Corticosteroid therapy for relapse results in
diuresis within one-week, enabling loss of retained
extracellular fluid [97,101]. Patients are advised to limit
sodium intake (1-2 mEq/kg/day; 15-35 mg/kg salt). Foods rich in
salt (>10 mEq/100 g; e.g., bread, cornflakes, processed cheese,
sauces, potato chips, salted nuts, papad, pickles) and
preserved foods (canned vegetables, soups and meat) are avoided
in presence of significant edema [97,101].
Diuretics are the initial therapy for
patients who are volume replete. Patients with moderate edema
without hypovolemia are managed with furosemide (2-4 mg/kg/day)
that acts on the ascending limb of Henle [101,105]. Sequential
nephron blockade, with additional use of hydrochlorothiazide
(2-4 mg/kg/day) or metolazone (0.1-0.2 mg/kg q12-24 hr),
augments diuresis by reducing distal sodium reabsorption
[97,101]. Monitoring for hypovolemia, hypokalemia and alkalosis
is essential. Spironolactone has limited diuretic efficacy, but
is an effective potassium-sparing agent in patients receiving
high-dose furosemide [97,101]. Use of acetazolamide or amiloride
is not advised.
Patients with severe edema may fail to
respond to maximal doses of furosemide and thiazide diuretics
(diuretic resistance) [98]. Factors contributing to diuretic
resistance are poor adherence to salt restriction, reduced
bioavailability of furosemide, hypoalbuminemia, hypovolemia, and
compensatory salt reabsorption in the distal tubule. The
bioavailability of oral furosemide is 20-60%, and is impaired by
gut edema in nephrotic syndrome [98]. In patients unresponsive
to oral furosemide, assessed as absence of diuresis within 2-4
hr of its administration, switching to IV therapy may elicit a
response. IV furosemide, given either as 1-2 mg/kg q 8-12 hr, or
bolus of 1 mg/kg followed by infusion of 0.1-0.4 mg/kg/hr is
effective [97,98,101]. While torsemide has better efficacy and
bioavailability than furosemide in adults with heart failure
[111], information in nephrotic syndrome is lacking.
Furosemide, tightly bound to blood albumin,
is actively secreted via organic acid pumps in the
ascending limb of Henle. Tubular secretion is impaired in
patients with severe hypoalbuminemia, resulting in diuretic
resistance [101]. The combination of 20% albumin (0.5-1 g/kg
infused over 3-4 hr) and furosemide (1-2 mg/kg at end of
infusion) enhances drug delivery to tubules, with increased
efficacy in terms of urine output and weight loss [110,112,113].
A meta-analysis confirmed that combination therapy results in
diuresis and natriuresis, which declines by 24-hr [101,114].
Therapy with IV albumin may be associated with risk of worsening
hypertension, respiratory distress and heart failure, and is
therefore avoided in patients with impaired kidney function
[97-99,101,112].
Patients with severe edema who are refractory
to the above therapies are likely to have fluid overload,
usually in presence of steroid resistance or kidney dysfunction.
These patients might require ultrafiltration or kidney
replacement therapy. An approach to evaluation and management of
edema is shown in Fig. 2.
Guideline 7: Infections and Immunization
7.1 Bacterial infections
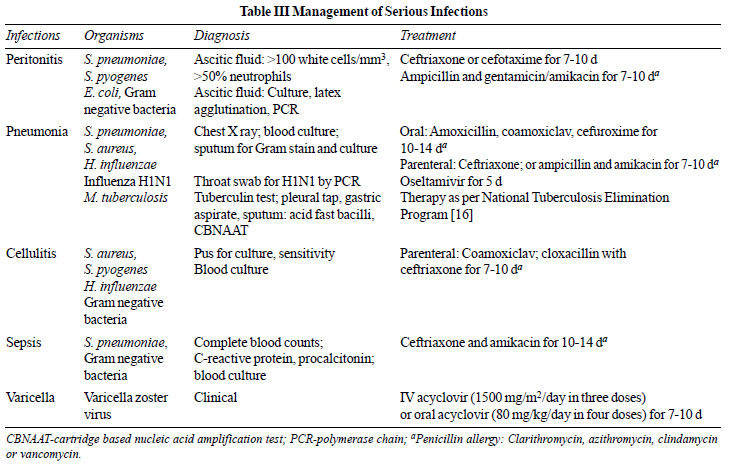
We suggest that serious bacterial infections
associated with nephrotic syndrome be managed as indicated in
Table III. (X)
Rationale
Infections are the chief complication in
patients with SSNS, accounting for 19-44% of hospitalizations
[115-120]. Contributing factors include the use of
immunosuppressive agents, anasarca, and urinary losses of IgG
and complement factors, that predispose to infection with
encapsulated organisms [121]. Peritonitis is the most common
severe infection, followed by pneumonia and cellulitis
[115-119]. Chief pathogens causing peritonitis are pneumococci
and E. coli; those causing pneumonia include pneumococci,
H. influenzae and S. aureus; and those responsible
for cellulitis are staphylococci, group A streptococci and H.
influenzae [115-119]. The diagnosis and treatment of severe
infections should follow standard guidelines [122-124] (Table
III). Apart from vaccines, there is no evidence of
efficacy of other interventions for preventing bacterial
infections in patients with nephrotic syndrome [125].
Viral infections
Several viruses, including rhinovirus,
adenovirus, influenza, parainfluenza, enterovirus, and
respiratory syncytial and Epstein Barr viruses, might trigger
disease relapses [126,127]. Infections such as varicella, zoster
and influenza might be associated with significant morbidity,
and merit specific prevention and management [128-130].
Severe acute respiratory syndrome
coronavirus-2 (SARS-CoV-2) infection:
Infection with SARS-CoV2, the
etiological agent of coronavirus disease (COVID-19), poses
challenges in management of patients with nephrotic syndrome
[131]. While children show mild disease, patients on
immunosuppression constitute a high-risk group that is
predisposed to adverse outcomes. Affected patients are at risk
of AKI, particularly if associated with hypovolemia or
aggressive use of diuretics. In absence of specific therapy for
SARS-CoV-2 infection, most expert groups advise reduction of
immunosuppression to acceptable levels, balancing the risk of
disease relapses against infection [131,132]. Other
considerations include advice through teleconsultation; low
threshold for inpatient monitoring of infected patients; and
limiting the use of biological agents and antimetabolites
[131,132]. Steroid dosing during SARS-CoV-2 infection should
follow standard practices regarding stress dosing [10]; relapses
may be treated with a lower dose of prednisolone.
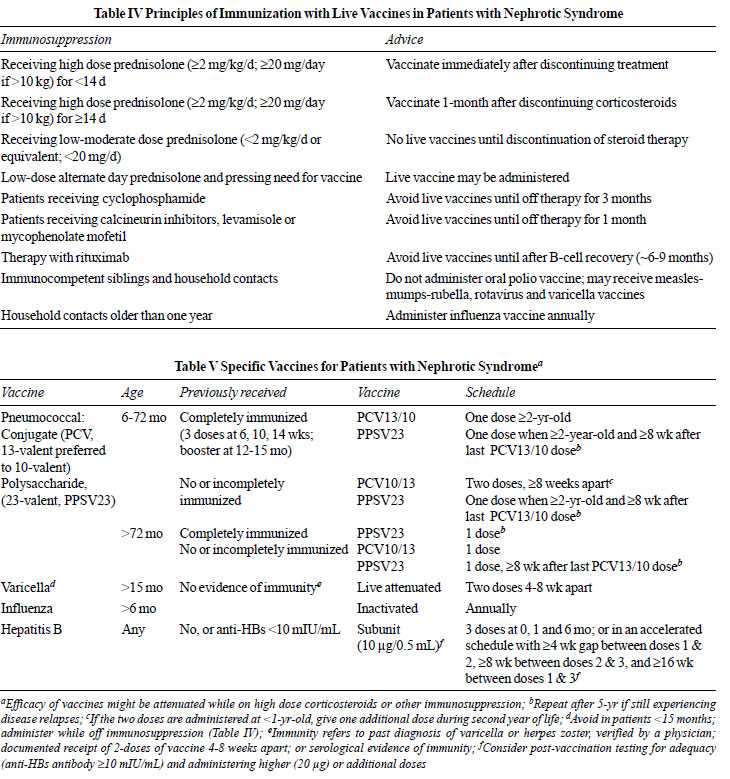
7.2 Immunization
We suggest that patients with nephrotic
syndrome receive: (i) age-appropriate killed, subunit or
inactivated vaccines; (ii) live vaccines following
principles outlined in Table IV; (iii) vaccines
against pneumococcus, varicella, influenza and hepatitis B (Table V).
(X)
Rationale
Children with nephrotic syndrome should
receive vaccines as appropriate for age [133,134]. Killed,
inactivated or subunit vaccines are not contraindicated, but may
have reduced efficacy during immunosuppression [133-136].
Principles of immunization with live vaccines in
immunocompromised children and their household contacts are
listed in Table IV [124,134,137]. The schedule for
administration of specific vaccines that are relevant to
patients with nephrotic syndrome is summarized in Table
V [133,134,138]. The risk of relapse following
vaccination is negligible [135,139].
Pneumococcal Vaccine
The availability of safe and immunogenic
vaccines has reduced the risk of pneumococcal infections in
patients with relapsing nephrotic syndrome [140]. Two categories
of vaccines are available. The polysaccharide vaccine (PPSV23)
is poorly immunogenic in patients younger than 2-years, and does
not induce immunological memory. Conjugate vaccines (PCV7-, 10-
and 13-valent) induce superior and sustained antibody responses
and immune memory even in young infants, with pooled efficacy of
58% (95% CI 29-75%) against invasive disease caused by any
pneumococcal serotype [135,141]. The efficacy of PPSV23 and PCV
vaccines in patients with SSNS is variable. Information is
lacking on the precise impact of vaccination on rates of
peritonitis, cellulitis and pneumonia.
Both PCV7/10/13 and PPSV23 elicit
satisfactory serological response, even when given during
relapse or while on immunosuppressive agents [135].
Nevertheless, we suggest that the vaccine be preferably given
during remission, and while on low or no immunosuppression.
Antibody responses are ill-sustained in patients with recurrent
relapses, justifying re-dosing with PPVS23 after 5 years if the
disease remains active; more than 2-doses of PPSV23 are not
recommended [134,135].
Varicella Vaccine
In view of the risk of severe disease in
immunocompromised patients, we recommend that patients with
nephrotic syndrome receive two doses of the varicella vaccine,
4-8 weeks apart (Table V) [134,138]. Two doses
result in seroconversion in ~95% vaccinees; breakthrough
varicella might occur in 2.2-7.3% children [142]. The vaccine
was safe and immunogenic in 109 patients with nephrotic
syndrome, including those receiving low-dose corticosteroids, in
two prospective series [143,144] and in an open-label RCT [145].
Severe varicella might follow infection in
at-risk individuals exposed to persons with either varicella or
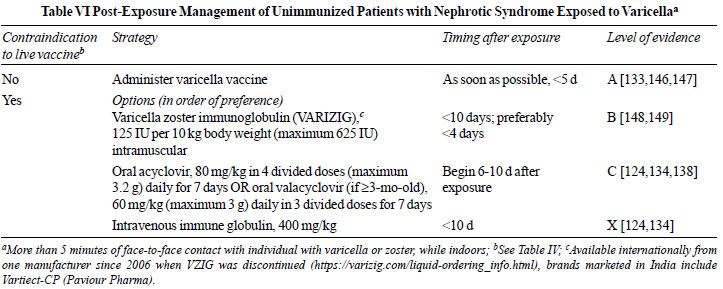
herpes zoster. Multiple strategies for post-exposure prophylaxis
are used to prevent viral transmission (Table VI)
[124,133,134,138,146-149]. Unimmunized patients with nephrotic
syndrome who are not immunosuppressed should receive the vaccine
within 5-days of exposure [124]. The risk of post-exposure
varicella was reduced to one-third in children who were
vaccinated following exposure, compared to those unimmunized (3
studies; n=110; 23% vs. 78%) [147]. Healthy
household contacts should also receive the vaccine to minimize
the risk of infecting the patient. In patients in whom
vaccination is contraindicated, the Center for Disease Control
recommends administration of varicella zoster immune globulin
(VARIZIG) within 10-d of exposure [148]. VARIZIG administration
was associated with varicella in <10% of 507 high-risk
participants, including 231 immunosuppressed children [149]. In
view of the low and variable titer of anti-VZV antibodies [150],
intravenous immunoglobulin (IVIG) is not recommended [124,134].
If VARIZIG is not available, similar to guidelines from the
American Academy of Pediatrics [124] and French Society of
Pediatric Nephrology [138], we recommend administering oral
acyclovir or valacyclovir for 7-days, starting 6-10 days after
exposure, corresponding to the period of secondary viremia (Table
VI).
 |
Influenza Vaccine
Influenza accounts for 13% of all pneumonia,
and 7% of severe pneumonia in children <5-yr-old [150,151].
Approximately 1 in 5 unvaccinated children are annually infected
by influenza, of which one-half are symptomatic [152]. Given the
risk of morbidity in immunosuppressed individuals, annual
administration of the inactivated influenza vaccine is
recommended for patients with nephrotic syndrome (Table V),
and their household contacts [124,130,138].
Hepatitis B Vaccine
Hepatitis B vaccination coverage rates in
India are unsatisfactory, and 45% of 1-6 yr-old children are not
vaccinated [153]. Compared to healthy children, fewer patients
with nephrotic syndrome show seroprotective ( ³10
mIU/mL) antibody titers [154]; one-half of these patients
seroconvert after vaccination [136,155]. Seroprotection was
lower in patients with steroid resistance, and those on
non-steroid therapies [136,154,155]. To overcome vaccine
failure, we advise an accelerated schedule using twice the
age-appropriate dose, and assessment of serological response to
administer booster dose(s) (Table V) [156].
Guideline 8: Transition of care
We recommend that patients with nephrotic
syndrome who continue to have relapses in adolescence be
transitioned into care by adult physicians. (X)
Rationale
SSNS is a self-limiting illness, with the
majority of patients outgrowing the illness by puberty. Review
of information from multiple cohorts, with median follow-up of
4-30 yr, indicates that the frequency of relapses declines with
age [3,4,157-159]. However, 5-42% patients may continue to have
active disease in adulthood. Risk factors for illness persisting
beyond 18-yr of age include early age at onset, and frequently
relapsing or steroid dependent course [3,4,157,158].
Major infections, associated with relapses
and intense immunosuppression, are the chief cause of
hospitalization and mortality (0-8%) [3,157,158]. Kidney failure
is uncommon (<1%) in patients with SSNS. There is significant
risk of short stature (15%), obesity (10%), hypertension
(6-46%), metabolic bone disease (9-63%), diabetes mellitus (2%),
ocular complications (10%), infertility and malignancies
[157,158,160]. Psychosocial concerns, including school drop-out,
unemployment and unstable relationships are common [161].
Given the risk of disease persistence and
prevalence of complications, it is advised to transfer the care
of adolescents with relapsing disease to ‘adult’ nephro-logists
by 18 year of age. National and international guidelines
advocate for smooth transition, with emphasis on shared clinics
and consideration of patient and parent perspectives [162].
CONCLUSIONS
The present guidelines, based on best
available evidence and expert guidance, provide directions for
evaluation and management of SSNS in children. Recommendations,
proposed by the Indian Society of Pediatric Nephrology, in 2001
and 2008, have been revised based on systematic reviews,
published studies and expert opinion. The management of frequent
relapses continues to be challenging, with morbidities
associated with the disease as well as therapies. Well-designed
prospective studies are required to address issues related to
therapy of the initial episode and relapsing nephrotic syndrome
(Table VII). We hope that the present guidelines
will standardize therapies and improve the quality of care for
patients with the disease.
Note: Supplementary material
related to these recommedations is available with the online
version at www.indianpediatrics.net
Contributors: All authors were involved
in review of literature, preparation of background document,
drafting and critically revising the manuscript. All authors
approved the final version of the manuscript.
Funding: Indian Council of Medical
Research; Advanced Centre for Research in Pediatric Kidney
Diseases [5/7/1090/2013-RHN]; Department of Biotechnology,
Government of India [BT/PR11030/MED/30/1644/2016].
Competing interests: None stated.
Annexure I
Expert Group of Indian Society of Pediatric
Nephrology
Participants: Anil Vasudevan,
Bengaluru; Abhijeet Saha, New Delhi; Aditi Sinha,
New Delhi; Aliza Mittal, Jodhpur; Amarjeet Mehta,
Jaipur; Arpana Iyengar, Bengaluru; Arpita Gogoi,
Dibrugarh; Anand S Vasudev, New Delhi; Pankaj Hari,
New Delhi; Ranjeet Thergaonkar, Mumbai; Priyanka
Khandelwal, New Delhi; Girish C Bhatt, Bhopal;
Indira Agarwal, Vellore; Jitendra K Meena, New Delhi;
Jyoti Sharma, Pune; Kanika Kapoor, New Delhi;
Kamran Afzal, Aligarh; Kanav Anand, New Delhi;
Karalanglin Tiewsoh, Chandigarh; Kirtisudha Mishra,
New Delhi; M Ashraf, Srinagar; Manish Kumar, New
Delhi; Manisha Sahay, Hyderabad; Mukta Mantan, New
Delhi; OP Mishra, Varanasi; PK Pruthi, New Delhi;
Rajiv Sinha, Kolkata Shobha Sharma, New Delhi;
Subal Pradhan, Cuttack; Sudha Ekambaram, Chennai;
Susan Uthup, Thiruvananthapuram; Sanjeev Gulati, New
Delhi; Saroj K Patnaik, New Delhi; Sriram
Krishnamurthy, Puducherry; Suprita Kalra, New Delhi;
Sushmita Banerjee, Kolkata; Vinay Agarwal, New
Delhi; Sumantra Raut, Kolkata; Arvind Bagga, New
Delhi, India.
Experts: Uma Ali, Mumbai; Kumud Mehta, Mumbai;
Madhuri Kanitkar, New Delhi; Amit K Dinda, New Delhi;
Geetika Singh, New Delhi; Kishore D Phadke, Bengaluru;
BR Nammalwar, Chennai; RN Srivastava, New Delhi.
REFERENCES
1. Noone DG, Iijima K, Parekh R. Idiopathic
nephrotic syndrome in children. Lancet. 2018;392:61-74
2. Banh THM, Hussain-Shamsy N, Patel V, et
al. Ethnic differences in incidence and outcomes of childhood
nephrotic syndrome. Clin J Am Soc Nephrol. 2016;11:1760-8.
3. Tarshish P, Tobin JN, Bernstein J,
Edelmann CM Jr. Prognostic significance of the early course of
minimal change nephrotic syndrome: Report of the International
Study of Kidney Disease in Children. J Am Soc Nephrol.
1997;8:769-76.
4. Sinha A, Hari P, Sharma PK, et al. Disease
course in steroid sensitive nephrotic syndrome. Indian Pediatr.
2012;49:881-7.
5. Kim JS, Bellew CA, Silverstein DM, et al.
High incidence of initial and late steroid resistance in
childhood nephrotic syndrome. Kidney Int. 2005;68:1275-81.
6. Indian Pediatric Nephrology Group, Indian
Academy of Pediatrics. Consensus Statement on Management of
Steroid Sensitive Nephrotic Syndrome. Indian Pediatr
2001;38:975-86.
7. Indian Pediatric Nephrology Group, Indian
Academy of Pediatrics, Bagga A, Ali U, Banerjee S, Kanitkar M,
Phadke KD, Senguttuvan P, Sethi S, Shah M. Management of Steroid
Sensitive Nephrotic Syndrome: Revised Guidelines. Indian Pediatr.
2008;45:203-14.
8. Guyatt GH, Oxman AD, Vist GE, et al; GRADE
Working Group. GRADE: An emerging consensus on rating quality of
evidence and strength of recommendations. BMJ. 2008;336:924-6.
9. Kidney Disease Improving Global Outcomes
Expert Group on Glomerular Diseases. KDIGO Clinical Practice
Guideline on Glomerular Diseases: Public Review draft. Accessed
June 15, 2020. Available from https://kdigo.org/guidelines/gn/
10. Indian Society of Pediatric Nephrology.
Revised Guidelines on Management of Steroid Resistant Nephrotic
Syndrome. Indian Pediatr. 2021:S097475591600278 (online ahead of
print).
11. American Academy of Pediatrics Steering
Committee on Quality Improvement and Management. Classifying
Recommendations for Clinical Practice Guidelines. Pediatrics.
2004;114:874-7.
12. Petersmann A, Müller-Wieland D, Müller
UA, et al. Definition, classification and diagnosis of diabetes
mellitus. Exp Clin Endocrinol Diabetes. 2019;127:S1-S7.
13. Khadilkar VV, Khadilkar AV. Revised
Indian Academy of Pediatrics 2015 growth charts for height,
weight and body mass index for 5-18-year-old Indian children.
Indian J Endocrinol Metab. 2015;19:470-6.
14. Khadilkar V, Khadilkar A, Arya A, et al.
Height velocity percentiles in Indian children aged 5–17 years.
Indian Pediatr. 2019;56:23-8.
15. Sathiyamoorthy R, Kalaivani M, Aggarwal
P, Gupta SK. Prevalence of pulmonary tuberculosis in India: A
systematic review and meta-analysis. Lung India. 2020;37:45-52.
16. National Strategic Plan for Tuberculosis
Elimination 2017-2025. Available from:
https://tbcindia.gov.in/WriteReadData/NSP%20Draft%2020.02.2017%201.pdf.
Accessed November 1, 2020.
17. Batham A, Narula D, Toteja T, et al.
Systematic review and meta-analysis of prevalence of hepatitis B
in India. Indian Pediatr. 2007;44:663-74.
18. Lane BM, Cason R, Esezobor CI, Gbadegesin
RA. Genetics of childhood steroid sensitive nephrotic syndrome:
An update. Front Pediatr. 2019;7:8.
19. Uwaezuoke SN. The role of novel
biomarkers in childhood idiopathic nephrotic syndrome: A
narrative review of published evidence. Int J Nephrol Renovasc
Dis. 2017;10:123-8.
20. Nephrotic syndrome in children:
Prediction of histopathology from clinical and laboratory
characteristics at time of diagnosis. A report of the
International Study of Kidney Disease in Children. Kidney Int.
1978;13:159-65.
21. White RH, Glasgow EF, Mills RJ.
Clinicopathological study of nephrotic syndrome in
childhood. Lancet. 1970;1:1353-9.
22. Srivastava RN, Mayekar G, Anand R,
Choudhry VP, Ghai OP, Tandon HD. Nephrotic syndrome in Indian
children. Arch Dis Child. 1975;50:626-30.
23. Gipson DS, Massengill SF, Yao L, et al.
Management of childhood onset nephrotic syndrome. Pediatrics.
2009;124:747-57.
24. Trautmann A, Vivarelli M, Samuel S, et
al; International Pediatric Nephrology Association: IPNA
Clinical Practice Recommendations for the Diagnosis and
Management of Children with Steroid Resistant Nephrotic
Syndrome. Pediatr Nephrol. 2020;35:1529-61.
25. Alshami A, Roshan A, Catapang M, et al;
Pediatric Nephrology Clinical Pathway Development Team.
Indications for kidney biopsy in idiopathic childhood nephrotic
syndrome. Pediatr Nephrol. 2017;32:1897-905.
26. Rutjes N, Sinha A, Bagga A, et al.
Outcome of steroid sensitive idiopathic nephrotic syndrome
commencing after the age of 12 years. 45th Annual Meeting of the
European Society of Pediatric Nephrology, Krakow, Poland.
Pediatr Nephrol. 2012;27:1704.
27. Gulati S, Sural S, Sharma RK, et al.
Spectrum of adolescent-onset nephrotic syndrome in Indian
children. Pediatr Nephrol. 2001;16:1045-8.
28. Webb NJ, Lewis MA, Iqbal J, Smart PJ,
Lendon M, Postlethwaite RJ. Childhood steroid sensitive
nephrotic syndrome: Does the histology matter? Am J Kidney Dis.
1996;27:484-8.
29. KDIGO Clinical Practice Guidelines.
General principles in management of glomerular disease. KI
Suppl. 2012;S2:S156-62.
30. Ishikura K, Matsumoto S, Sako M, et al;
Japanese Society for Pediatric Nephrology. Clinical practice
guideline for pediatric idiopathic nephrotic syndrome 2013:
Medical therapy. Clin Exp Nephrol 2015;19:6-33.
31. Lusco MA, Fogo AB, Najafian B, Alpers CE.
AJKD atlas of renal pathology: Calcineurin inhibitor
nephrotoxicity. Am J Kidney Dis. 2017;69:e21-2.
32. Liu F, Mao JH. Calcineurin inhibitors and
nephrotoxicity in children. World J Pediatr. 2018;14:121-6.
33. Iijima K, Hamahira K, Tanaka R, et al.
Risk factors for cyclosporine-induced tubulointerstitial lesions
in children with minimal change nephrotic syndrome. Kidney Int.
2002;61:1801-5.
34. Sinha A, Sharma A, Mehta A, et al.
Calcineurin inhibitor induced nephrotoxicity in steroid
resistant nephrotic syndrome. Indian J Nephrol. 2013;23:41-6.
35. Fujinaga S, Kaneko K, Muto T, et al.
Independent risk factors for chronic cyclosporine induced
nephropathy in children with nephrotic syndrome. Arch Dis Child.
2006;91:666-70.
36. Delbet JD, Aoun B, Buob D, et al.
Infrequent tacrolimus-induced nephrotoxicity in French patients
with steroid-dependent nephrotic syndrome. Pediatr Nephrol.
2019;34:2605-8.
37. Nankivell BJ, Ng CH, O Connell PJ,
Chapman JR. Calcineurin inhibitor nephrotoxicity through the
lens of longitudinal histology: Comparison of cyclosporine and
tacrolimus eras. Transplantation. 2016;100:1723-31.
38. Ishikura K, Yoshikawa N, Hattori S, et
al; for Japanese Study Group of Renal Disease in Children.
Treatment with microemulsified cyclosporine in children with
frequently relapsing nephrotic syndrome. Nephrol Dial
Transplant. 2010;25:3956-62.
39. Corwin HL, Schwartz MM, Lewis EJ. The
importance of sample size in the interpretation of the renal
biopsy. Am J Nephrol. 1988;8:85-9.
40. Pavlisko EN, Howell DN. The continued
vital role of electron microscopy in the diagnosis of renal
disease/dysfunction. Ultrastruct Pathol. 2013;37:1-8.
41. The primary nephrotic syndrome in
children. Identification of patients with minimal change
nephrotic syndrome from initial response to prednisolone. A
report of the International Study of Kidney Disease in Children.
J Pediatr. 1981;98:556-64.
42. Ehrich JH, Brodehl J. Long versus
standard prednisone therapy for initial treatment of idiopathic
nephrotic syndrome in children. Arbeitsgemeinschaft fur
Padiatrische Nephrologie. Eur J Pediatr. 1993;152:357-61.
43. Hodson EM, Willis NS, Craig JC.
Corticosteroid therapy for nephrotic syndrome in children.
Cochrane Database Syst Rev. 2007;4:CD001533.
44. Hahn D, Hodson EM, Willis NS, Craig JC.
Corticosteroid therapy for nephrotic syndrome in children.
Cochrane Database Syst Rev. 2015;3:CD001533.
45. Webb NJA, Woolley RL, Lambe T, et al;
PREDNOS Collaborative Group. Long term tapering versus standard
prednisolone treatment for first episode of childhood nephrotic
syndrome: Phase III randomized controlled trial and economic
evaluation. BMJ. 2019;365:l1800.
46. Sinha A, Saha A, Kumar M, et al.
Extending initial prednisolone treatment in a randomized control
trial from 3 to 6 months did not significantly influence the
course of illness in children with steroid-sensitive nephrotic
syndrome. Kidney Int. 2015;87:217-24.
47. Emma F, Montini G, Gargiulo A. Equations
to estimate prednisone dose using body weight. Pediatr Nephrol.
2019;34:685-8.
48. Redlarski G, Palkowski A, Krawczuk M.
Body surface area formulae: An alarming ambiguity. Sci Rep.
2016;6:27966.
49. Feber J, Al-Matrafi J, Farhadi E,
Vaillancourt R, Wolfish N. Prednisone dosing per body weight or
body surface area in children with nephrotic syndrome: Is it
equivalent? Pediatr Nephrol. 2009;24:1027-31.
50. Saadeh SA, Baracco R, Jain A, et al.
Weight or body surface area dosing of steroids in nephrotic
syndrome: Is there an outcome difference? Pediatr Nephrol.
2011;26:2167-71.
51. Hirano D, Fujinaga S. Two dosing regimens
for steroid therapy in nephrotic syndrome. Pediatr Nephrol.
2014;29:325.
52. Raman V, Krishnamurthy S,
Harichandrakumar KT. Body weight-based prednisolone versus body
surface area- based prednisolone for induction of remission in
children with nephrotic syndrome: a randomized, open–label,
equivalence clinical trial. Pediatr Nephrol. 2016;31:595-604.
53. Basu B, Bhattacharyya S, Barua S, Naskar
A, Roy B. Efficacy of body weight vs body surface area-based
prednisolone regimen in nephrotic syndrome. Clin Exp Nephrol.
2020;24:622-9.
54. Ekka BK, Bagga A, Srivastava RN. Single-
versus divided-dose prednisolone therapy for relapses of
nephrotic syndrome. Pediatr Nephrol. 1997;11:597-9.
55. Schijvens AM, Ter Heine R, de Wildt SN,
Schreuder MF. Pharmacology and pharmacogenetics of prednisone
and prednisolone in patients with nephrotic syndrome. Pediatr
Nephrol. 2019;34:389-403.
56. Aljebab F, Choonara I, Conroy S.
Systematic review of the toxicity of short-course oral
corticosteroids in children. Arch Dis Child. 2016;101:365-70.
57. Liu D, Ahmet A, Ward L, et al. A
practical guide to the monitoring and management of the
complications of systemic corticosteroid therapy. Allergy Asthma
Clin Immunol. 2013;9:30.
58. Lombel RM, Gipson DS, Hodson EM; Kidney
Disease: Improving Global Outcomes. Treatment of
Steroid-Sensitive Nephrotic Syndrome: New Guidelines from KDIGO.
Pediatr Nephrol. 2013;28:415-26.
59. Abeyagunawardena AS, Thalgahagoda RS,
Dissanayake PV, et al. Short courses of daily prednisolone
during upper respiratory tract infections reduce relapse
frequency in childhood nephrotic syndrome. Pediatr Nephrol.
2017;32:1377-82.
60. Yadav M, Sinha A, Khandelwal P, Hari P,
Bagga A. Efficacy of low-dose daily versus alternate-day
prednisolone in frequently relapsing nephrotic syndrome: An
open-label randomized controlled trial. Pediatr Nephrol.
2019;34: 829-35.
61. Srivastava RN, Vasudev AS, Bagga A,
Sunderam KR. Long-term, low-dose prednisolone therapy in
frequently relapsing nephrotic syndrome. Pediatr Nephrol.
1992;6:247-50.
62. Hodson EM, Craig JC. In steroid sensitive
nephrotic syndrome in children, is there clear evidence that
steroids given every second day are more beneficial in terms of
reducing relapse rate and side effects compared with half the
dose given every day? Pediatr Nephrol. 2001;16:1159-60.
63. Mühlig AK, Lee JY, Kemper MJ, et al.
Levamisole in children with idiopathic nephrotic syndrome:
Clinical efficacy and pathophysiological aspects. J Clin Med.
2019;8:860.
64. Larkins NG, Liu ID, Willis NS, Craig JC,
Hodson EM. Non-corticosteroid immunosuppressive medications for
steroid-sensitive nephrotic syndrome in children. Cochrane
Database Syst Rev. 2020;4:CD002290
65. Gruppen MP, Bouts AH, Jansen-van der
Weide MC, et al; members of the Levamisole Study Group. A
randomized clinical trial indicates that levamisole increases
the time to relapse in children with steroid-sensitive
idiopathic nephrotic syndrome. Kidney Int. 2018;93: 510-8.
66. Vivarelli M, Emma F. Levamisole for
children with nephrotic syndrome: New evidence for the use of an
"old" drug. Kidney Int. 2019;95:25-8.
67. Jin Q, Kant S, Alhariri J, Geetha D.
Levamisole adulterated cocaine associated ANCA vasculitis:
Review of literature and update on pathogenesis. J Community
Hosp Intern Med Perspect. 2018;8: 339-44.
68. Querfeld U, Weber LT. Mycophenolate
mofetil for sustained remission in nephrotic syndrome. Pediatr
Nephrol. 2018;33: 2253-65.
69. Jellouli M, Fitouhi S, Abidi K, et al.
Mycophenolate mofetil in treatment of childhood
steroid-dependent nephrotic syndrome. Tunis Med. 2016;94:221-5.
70. Sinha A, Puraswani M, Kalaivani M, Goyal
P, Hari P, Bagga A. Efficacy and safety of mycophenolate mofetil
versus levamisole in frequently relapsing nephrotic syndrome: An
open-labelrandomized controlled trial. Kidney Int.
2019;95:210-8.
71. Gellermann J, Weber L, Pape L, Tonshoff
B, Hoyer P, Querfeld U. Mycophenolate mofetil versus cyclosporin
A in children with frequently relapsing nephrotic syndrome. J Am
Soc Nephrol. 2013; 24:1689-97.
72. Tong K, Mao J, Fu H, et al. The value of
monitoring the serum concentration of mycophenolate mofetil in
children with steroid-dependent/frequent relapsing nephrotic
syndrome. Nephron. 2016;132:327-34.
73. Hackl A, Cseprekal O, Gessner M, et al.
Mycophenolate mofetil therapy in children with idiopathic
nephrotic syndrome: Does therapeutic drug monitoring make a
difference? Ther Drug Monit. 2016;38:274-9.
74. Sobiak J, Resztak M, Ostalska-Nowicka D,
et al. Monitoring of mycophenolate mofetil metabolites in
children with nephrotic syndrome and the proposed novel target
values of pharmacokinetic parameters. Eur J Pharm Sci.
2015;77:189-96.
75. Mendizábal S, Zamora I, Berbel O, et al.
Mycophenolate mofetil in
steroid/cyclosporine-dependent/resistant nephrotic syndrome.
Pediatr Nephrol. 2005;20:914-9.
76. Fujinaga S, Ohtomo Y, Umino D, et al. A
prospective study on the use of mycophenolate mofetil in
children with cyclosporine-dependent nephrotic syndrome. Pediatr
Nephrol. 2007;22:71-6.
77. Fujinaga S, Ohtomo Y, Hirano D, et al.
Mycophenolate mofetil therapy for childhood-onset steroid
dependent nephrotic syndrome after long-term cyclosporine:
extended experience in a single center. Clin Nephrol.
2009;72:268-73.
78. Benz MR, Ehren R, Kleinert D, et al.
Generation and validation of a limited sampling strategy to
monitor mycophenolic acid exposure in children with nephrotic
syndrome. Ther Drug Monit. 2019;41: 696-702.
79. Latta K, von Schnakenburg C, Ehrich JH. A
meta-analysis of cytotoxic treatment for frequently relapsing
nephrotic syndrome in children. Pediatr Nephrol. 2001;16:271-82.
80. Fu HD, Qian GL, Jiang ZY. Comparison of
second-line immunosuppressants for childhood refractory
nephrotic syndrome: A systematic review and network
meta-analysis. J Investig Med. 2017;65:65-71.
81. Rivkees SA, Crawford JD. The relationship
of gonadal activity and chemotherapy-induced gonadal damage.
JAMA. 1988;259:2123-5.
82. Arslansoyu Camlar S, Soylu A, Kavukçu S.
Cyclosporine in pediatric nephrology. Iran J Kidney Dis.
2018;12:319-30.
83. Hao GX, Song LL, Zhang DF, Su LQ,
Jacqz-Aigrain E, Zhao W. Off-label use of tacrolimus in children
with glomerular disease: Effectiveness, safety and
pharmacokinetics. Br J Clin Pharmacol. 2020;86:274-84.
84. Sinha A, Bagga A, Gulati A, Hari P.
Short-term efficacy of rituximab versus tacrolimus in
steroid-dependent nephrotic syndrome. Pediatr Nephrol.
2012;27:235-41.
85. Fujinaga S, Ohtomo Y, Someya T, et al. Is
single-daily low-dose cyclosporine therapy really effective in
children with idiopathic frequent-relapsing nephrotic syndrome?
Clin Nephrol. 2008;69: 84-9.
86. Nakahata T, Tanaka H, Tsugawa K, et al.
C1-C2 point monitoring of low-dose cyclosporin a given as a
single daily dose in children with steroid-dependent relapsing
nephrotic syndrome. Clin Nephrol. 2005;64:258-63.
87. Sinha A, Bagga A. Rituximab therapy in
nephrotic syndrome: Implications for patients’ management. Nat
Rev Nephrol. 2013;9:154-69.
88. Kronbichler A, Kerschbaum J,
Fernandez-Fresnedo G, et al. Rituximab treatment for relapsing
minimal change disease and focal segmental glomerulosclerosis: A
systematic review. Am J Nephrol. 2014;39:322-30.
89. Sinha A, Bhatia D, Gulati A, et al.
Efficacy and safety of rituximab in children with
difficult-to-treat nephrotic syndrome. Nephrol Dial Transplant.
2015;30:96-106.
90. Jacobs R, Langer-Jacobus T, Duong M, et
al. Detection and quantification of rituximab in the human
urine. J Immunol Methods. 2017;451:118-21.
91. Chan EY, Webb H, Yu E, et al. Both the
rituximab dose and maintenance immunosuppression in
steroid-dependent/frequently-relapsing nephrotic syndrome have
important effects on outcomes. Kidney Int. 2020;97:393-401.
92. Tony HP, Burmester G, Schulze-Koops H, et
al; GRAID investigators. Safety and clinical outcomes of
rituximab therapy in patients with different autoimmune
diseases: Experience from a national registry (GRAID). Arthritis
Res Ther. 2011;13:R75.
93. Tang Z, Li X, Wu S, et al. Risk of
hepatitis B reactivation in HBsAg-negative/HBcAb-positive
patients with undetectable serum HBV DNA after treatment with
rituximab for lymphoma: A meta-analysis. Hepatol Int.
2017;11:429-33.
94. Khojah AM, Miller ML, Klein-Gitelman MS,
et al. Rituximab-associated hypogammaglobulinemia in pediatric
patients with autoimmune diseases. Pediatr Rheumatol Online J.
2019;17:61.
95. Parmentier C, Delbet JD, Decramer S, et
al. Immunoglobulin serum levels in rituximab-treated patients
with steroid-dependent nephrotic syndrome. Pediatr Nephrol.
2020;35:455-62.
96. Colucci M, Carsetti R, Serafinelli J, et
al. Prolonged impairment of immunological memory after anti-CD20
treatment in pediatric idiopathic nephrotic syndrome. Front
Immunol. 2019;10:1653.
97. Kaku Y, Ohtsuka Y, Komatsu Y, et al;
Japanese Society for Pediatric Nephrology. Clinical Practice
Guideline for Pediatric Idiopathic Nephrotic Syndrome 2013:
General Therapy. Clin Exp Nephrol. 2015;19:34-53.
98. Hoorn EJ, Ellison DH. Diuretic
resistance. Amer J Kidney Dis. 2017;69:136-42.
99. Büyükavci MA, Çivilibal M, Elevli M,
Selçuk Duru HN. Hypo- and hypervolemic edema in children with
steroid sensitive nephrotic syndrome. Turk J Med Sci.
2015;45:178-83.
100. Gurgoze MK, Gunduz Z, Poyrazoglu MH,
Dursun I, Uzum K, Dusunsel R. Role of sodium during formation of
edema in children with nephrotic syndrome. Pediatr Int.
2011;53:50-6.
101. Meena J, Bagga A. Current perspectives
in management of edema in nephrotic syndrome. Indian J Pediatr.
2020;87:633-40.
102. Marzuillo P, Guarino S, Apicella A, et
al. Assessment of volume status and appropriate fluid
replenishment in the setting of nephrotic syndrome. J Emerg Med.
2017;52:e149-52.
103. Keenswijk W, Ilias MI, Raes A, et al.
Urinary potassium to urinary potassium plus sodium ratio can
accurately identify hypovolemia in nephrotic syndrome: A
provisional study. Eur J Pediatr. 2018;177:79-8.
104. Matsumoto H, Miyaoka Y, Okada T, et al.
Ratio of urinary potassium to urinary sodium and the potassium
and edema status in nephrotic syndrome. Intern Med.
2011;50:551-5.
105. Kapur G, Valentini RP, Imam AA, Mattoo
TK. Treatment of severe edema in children with nephrotic
syndrome with diuretics alone: A prospective study. Clin J Am
Soc Nephrol. 2009;4:907-13.
106. Ray EC, Rondon-Berrios H, Boyd CR,
Kleyman TR. Sodium retention and volume expansion in nephrotic
syndrome: implications for hypertension. Adv Chronic Kidney Dis.
2015;22:179-84.
107. Hinrichs GR, Jensen BL, Svenningsen P.
Mechanisms of sodium retention in nephrotic syndrome. Curr Opin
Nephrol Hypertens. 2020;29:207-12.
108. Park SJ, Shin JI. Complications of
nephrotic syndrome. Korean J Pediatr. 2011;54:322-8.
109. Nalcacioglu H, Ozkaya O, Baysal K, et
al. The role of bioelectrical impedance analysis, NT-ProBNP and
inferior vena cava sonography in the assessment of body fluid
volume in children with nephrotic syndrome. Nefrologia.
2018;38:48-56.
110. Taneja K, Kumar V, Anand R, Pemde HK.
Normative data for IVC diameter and its correlation with the
somatic parameters in healthy Indian children. Indian J Pediatr.
2018;85:108-12.
111. Abraham B, Megaly M, Sous M, et al.
Meta-analysis comparing torsemide versus furosemide in patients
with heart failure. Am J Cardiol. 2020;125:92-9.
112.Elwell RJ, Spencer AP, Eisele G. Combined
furosemide and human albumin treatment for diuretic-resistant
edema. Ann Pharmacother. 2003;37:695-700.
113. Dharmaraj R, Hari P, Bagga A. Randomized
cross-over trial comparing albumin and frusemide infusions in
nephrotic syndrome. Pediatr Nephrol. 2009;24:775-82.
114. Ho JJ, Adnan AS, Kueh YC, Ambak NJ, Van
Rostenberghe H, Jummaat F. Human albumin infusion for treating
edema in people with nephrotic syndrome. Cochrane Database Syst
Rev. 2019;7:CD009692.
115. Alfakeekh K, Azar M, Sowailmi BA, et al.
Immunosuppressive burden and risk factors of infection in
primary childhood nephrotic syndrome. J Infect Public Health.
2019;12:90-4.
116. Mantan M, Singh S. Infection associated
relapses in children with nephrotic syndrome: A short-term
outcome study. Saudi J Kidney Dis Transpl. 2019;30:1245-53.
117. Wei CC, Yu IW, Lin HW, Tsai AC.
Occurrence of infection among children with nephrotic syndrome
during hospitalizations. Nephrology (Carlton). 2012;17:681-8.
118. Kumar M, Ghunawat J, Saikia D, Manchanda
V. Incidence and risk factors for major infections in
hospitalized children with nephrotic syndrome. J Bras Nefrol.
2019; 41:526-33.
119. Ajayan P, Krishnamurthy S, Biswal N,
Mandal J. Clinical spectrum and predictive risk factors of major
infections in hospitalized children with nephrotic syndrome.
Indian Pediatr. 2013;50:779-81.
120. Yamamoto R, Imai E, Maruyama S, et al.
Incidence of remission and relapse of proteinuria, end-stage
kidney disease, mortality, and major outcomes in primary
nephrotic syndrome: The Japan Nephrotic Syndrome Cohort Study
(JNSCS). Clin Exp Nephrol. 2020;24:526-40.
121. Sadarangani M. Protection against
invasive infections in children caused by encapsulated bacteria.
Front Immunol. 2018;9:2674.
122. Solomkin JS, Mazuski JE, Bradley JS, et
al. Diagnosis and management of complicated intra-abdominal
infection in adults and children: Guidelines by the Surgical
Infection Society and the Infectious Diseases Society of
America. Clin Infect Dis. 2010;50:133-6
123. Shriner A, Wilkie L. Pediatric
cellulitis: A red-hot concern. Pediatr Ann. 2017;46:e265-9.
124. Kimberlin DW, Brady MT, Jackson MA, Long
SS (Eds.). Red Book: 2018 Report of the Committee on Infectious
Diseases (31st ed.), American Academy of Pediatrics, Itasca, IL.
Accessed October 2, 2020. Available from:
http://search.ebscohost.com/login.aspx? direct=true&scope=site&db=nlebk&db=
nlabk&AN= 1809323
125. Wu HM, Tang JL, Cao L, Sha ZH, Li Y.
Interventions for preventing infection in nephrotic syndrome.
Cochrane Database Syst Rev. 2012;2012:CD003964.
126. MacDonald NE, Wolfish N, McLaine P,
Phipps P, Rossier E. Role of respiratory viruses in
exacerbations of primary nephrotic syndrome. J Pediatr.
1986;108:378-82.
127. Dossier C, Sellier-Leclerc AL, Rousseau
A, et al. Prevalence of herpesviruses at onset of idiopathic
nephrotic syndrome. Pediatr Nephrol. 2014;29:2325-31.
128. Leuvenink R, Aeschlimann F, Baer W, et
al. Clinical course and therapeutic approach to varicella zoster
virus infection in children with rheumatic autoimmune diseases
under immunosuppression. Pediatr Rheumatol Online J. 2016;14:34.
129. Gershon AA, Breuer J, Cohen JI, et al.
Varicella zoster virus infection. Nat Rev Dis Primers.
2015;1:15016.
130. Committee on Infectious Diseases.
Recommendations for prevention and control of influenza in
children, 2019-2020. Pediatr. 2019;144:e20192478.
131. Kronbichler A, Gauckler P, Windpessl M,
et al. COVID-19: Implications for immunosuppression in kidney
disease and transplantation. Nat Rev Nephrol. 2020;16:365-7.
132. Vasudevan A, Mantan M, Krishnamurthy S,
et al; Indian Society of Pediatric Nephrology. Managing children
with renal diseases during COVID-19 pandemic. Indian Pediatr.
2020;57:641-51.
133. Balasubramanian S, Shah A, Pemde HK, et
al; IAP Advisory Committee on Vaccines and Immunization
Practices, 2018-19. Indian Academy of Pediatrics (IAP) Advisory
Committee on Vaccines and Immunization Practices (ACVIP)
Recommended Immunization Schedule (2018-19) and Update on
Immunization for Children Aged 0 through 18 Years. Indian
Pediatr. 2018;55: 1066-74.
134. Centers for Disease Control and
Prevention. Epidemiology and prevention of vaccine-preventable
diseases. Hamborsky J, Kroger A, Wolfe S, eds. 13th ed.
Washington D.C. Public Health Foundation, 2015; Available from:
https://www.cdc.gov/vaccines/pubs/pinkbook/index.html.
Accessed October 2, 2020.
135. Goonewardene ST, Tang C, Tan LT, et al.
Safety and efficacy of pneumococcal vaccination in pediatric
nephrotic syndrome. Front Pediatr. 2019;7:339.
136. Yýldýz N, Sever L, Kasapçopur Ö, Çullu
F, Arýsoy N, Çalýþkan S. Hepatitis B virus vaccination in
children with steroid sensitive nephrotic syndrome:
Immunogenicity and safety? Vaccine. 2013;31:3309-12.
137. Banerjee S, Dissanayake PV,
Abeyagunawardena AS. Vaccinations in children on
immunosuppressive medications for renal disease. Pediatr
Nephrol. 2016;31:1437-48.
138. Boyer O, Baudouin V, Bérard É, et al.
Vaccine recommendations for children with idiopathic nephrotic
syndrome. Nephrol Ther. 2020;16:177-83.
139. Ishimori S, Kamei K, Ando T, et al.
Influenza virus vaccination in children with nephrotic syndrome:
Insignificant risk of relapse. Clin Exp Nephrol.
2020;24:1069-76.
140. McIntyre P, Craig JC. Prevention of
serious bacterial infection in children with nephrotic syndrome.
J Paediatr Child Health. 1998;34:314-7.
141. Lucero MG, Dulalia VE, Nillos LT, et al.
Pneumococcal conjugate vaccines for preventing vaccine-type
invasive pneumococcal disease and X-ray defined pneumonia
in children less than two years of age. Cochrane Database Syst
Rev. 2009;2009:CD004977.
142. Newman AM, Jhaveri R. Myths and
misconceptions: Varicella-zoster virus exposure, infection
risks, complications, and treatments. Clin Ther.
2019;41:1816-22.
143. Alpay H, Yildiz N, Onar A, Temizer H,
Ozçay S. Varicella vaccination in children with
steroid-sensitive nephrotic syndrome. Pediatr Nephrol.
2002;17:181-3.
144. Kamei K, Miyairi I, Ishikura K, et al.
Prospective study of live attenuated vaccines for patients with
nephrotic syndrome receiving immunosuppressive agents. J Pediatr.
2018;196:217-22.e1.
145. Furth SL, Arbus GS, Hogg R, Tarver J,
Chan C, Fivush BA; Southwest Pediatric Nephrology Study Group.
Varicella vaccination in children with nephrotic syndrome: A
report of the Southwest Pediatric Nephrology Study Group. J
Pediatr. 2003;142:145-8.
146. Lachiewicz AM, Srinivas ML. Varicella-zoster
virus post-exposure management and prophylaxis: A review. Prev
Med Rep. 2019;16:101016.
147. Macartney K, Heywood A, McIntyre P.
Vaccines for post exposure prophylaxis against varicella
(chickenpox) in children and adults. Cochrane Database Syst Rev.
2014;6:CD001833.
148. Centers for Disease Control and
Prevention. Updated recommendations for use of VariZIG-United
States, 2013. Morb Mortal Wkly Rep. 2013;62:574-6.
149. Levin MJ, Duchon JM, Swamy GK, Gershon
AA. Varicella zoster immune globulin (VARIZIG) administration up
to 10 d after varicella exposure in pregnant women,
immunocompromised participants, and infants: Varicella outcomes
and safety results from a large, open-label, expanded-access
program. PLoS One. 2019;14:e0217749.
150. Paryani SG, Arvin AM, Koropchak CM, et
al. Comparison of varicella zoster antibody titers in patients
given intravenous immune serum globulin or varicella zoster
immune globulin. J Pediatr. 1984;105:200-5.
151. Malosh RE, Martin ET, Ortiz JR, Monto
AS. The risk of lower respiratory tract infection following
influenza virus infection: A systematic and narrative review.
Vaccine. 2018;36:141-7.
152. Somes MP, Turner RM, Dwyer LJ, Newall
AT. Estimating the annual attack rate of seasonal influenza
among unvaccinated individuals: A systematic review and
meta-analysis. Vaccine. 2018;36:3199-207.
153. Khan J, Shil A, Mohanty SK. Hepatitis B
vaccination coverage across India: Exploring the spatial
heterogeneity and contextual determinants. BMC Public Health.
2019;19:1263.
154. Neupane N, Krishnamurthy S, Jagadisan B,
Dhodapkar R. Hepatitis B seroprotection in pediatric nephrotic
syndrome. Indian Pediatr. 2019;56:659-662.
155. Mantan M, Pandharikar N, Yadav S,
Chakravarti A, Sethi GR. Seroprotection for hepatitis B in
children with nephrotic syndrome. Pediatr Nephrol.
2013;28:2125-30.
156. Das S, Ramakrishnan K, Behera SK,
Ganesapandian M, Xavier AS, Selvarajan S. Hepatitis B vaccine
and immunoglobulin: Key concepts. J Clin Transl Hepatol.
2019;7:165-71.
157. Hjorten R, Anwar Z, Reidy KJ. Long-term
outcomes of childhood onset nephrotic syndrome. Front Pediatr.
2016;4:53.
158. Marchel DM, Gipson DS. Adult survivors
of idiopathic childhood onset nephrotic syndrome. Pediatr
Nephrol. 2020 Nov 6; DOI: 10.1007/s00467-020-04773-3 [Online
ahead of print]
159. Carter SA, Mistry S, Fitzpatrick J, et
al. Prediction of short- and long-term outcomes in childhood
nephrotic syndrome. Kidney Int Rep. 2019;5:426-34.
160. Lee JM, Kronbichler A, Shin JI, Oh J.
Review on long-term non-renal complications of childhood
nephrotic syndrome. Acta Paediatr. 2020;109:460-70.
161. Mehta M, Bagga A, Bajaj G, Srivastava
RN. Behavior problems in nephrotic syndrome. Indian Pediatr.
1995;32:1281-6.
162. Watson AR, Harden P, Ferris M, Kerr PG,
Mahan J, Ramzy MF. Transition from Pediatric to Adult Renal
Services: A Consensus Statement by the International Society of
Nephrology and the International Pediatric Nephrology
Association. Pediatr Nephrol. 2011;26:1753-7.
|
|
|
 |
|