|
A Child with a
DIC (15p; 22p) Centric Fusion and Fetal Valproate Syndrome |
N. Sharief
T. Adams*
L. butler*
J. gnannaratnam
From the Basildo(l General Hospital, Nether Mayne, Basildon, Essex,
U.K. and *North Thames (East) Regional Cytogenetic Unit, Institute of Neurology, Queen's Square, London, U. K.
Reprint requests: Dr. N. Sharief, Consultant Pediatrician, Basildon Hospital, Nether Mayne, Essex SSI65NL, U.K.
Manuscript Received: August 10,1998; Initial review completed: September 8, 1998; Revision Accepted: November 17, 1998
Centric fusion (Robertsonian Translocation), by definition occurs in acrocentric chromosomes with the break point close to the centromere. If the break points are located in the short arms near the centromere of both chromosomes, fusion of the two results in a dicentric chromosome (i.e., with two centromeres). A dicentric may consist of two nonhomologous chromosomes (asymmetric) or two homologous chromosomes (symmetric). Robertsonian translocation between chromosome IS and 22 have been described in the past but not in a dicentric form(1,2). Robertsonian fusions are not generally associated with specific clinical abnormalities but there may be a small risk of developmental delay if the anomaly is de novo.
The first report of Valproate embryopathy was published in 1980(3). It is now a well recognized syndrome including a distinctive facial phenotype, and other congenital mal- formations. Common facial features include
trigoncephaly, tall forehead with bifrontal narrowing, epicanthic folds, infraorbital groove, medial deficiency of eyebrows, a flat nasal bridge, broad nasal base, anteverted nostrils, shallow philtrum, long upper lip with thin vermilion border, thick lower lip and small down turned mouth(4-6). The most frequent malformations are neural tube defects, congenital heart disease, cleft lip and palate, genitourinary abnormalities, tracheomalacia, radial ray defects, abdominal wall defects and arachnodactyly/overlapping digits(6,7). Recently reduction deformity of limb(8), omphalocele(9), autism(10) and developmental delay(11) have all been reported in associa- tion with sodium valproate syndrome.
We describe a child with centric fusion between chromosome IS and 22 but whose clinical abnormalities are consistent with the exposure to sodium valproate throughout pregnancy.
Case Report
A male infant was born at 37 weeks gestation to a 26-year-old mother who suffers from epilepsy and she had been on Sodium valproate 500 mg twice daily at conception and throughout pregnancy. She has peroneal muscular atrophy type I and her own mother also suffers from the same condition. His Apgar scores were six at one minute and five at nine minutes. He required resuscitation with bag and mask for one minute. He was admitted to the neonatal unit because of
hypotherrnia and hypoglycemia but had no further problems. His birth weight was 2.02 kg and his head circumference 31.2 cm both below 3rd centile.
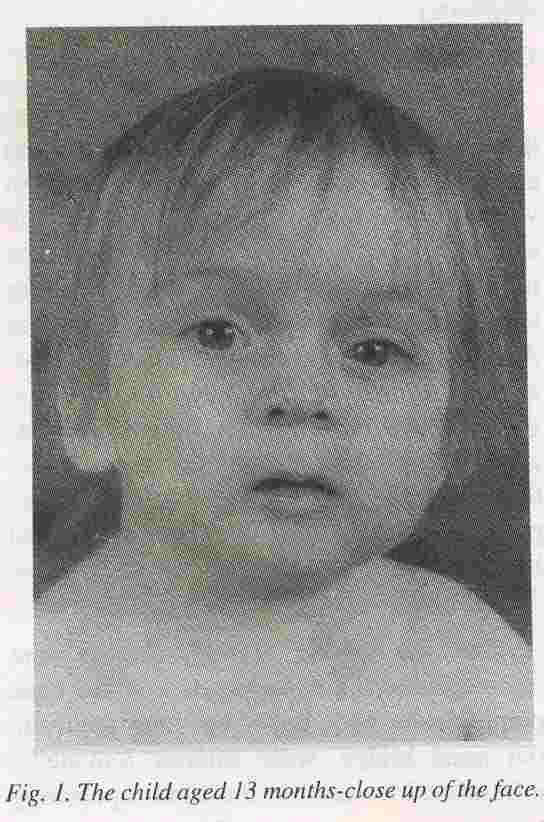
The dysmorphic features noted were trigoncephaly, high forehead, low set ears, pointed chin, wide upper lip, long philtrum, flat nasal bridge, wide anterior fontanelle, infra-orbital creases on the right, up slanting
palpebral fissures and mild hypertelorism (Fig. 1). His hands including the creases were normal. The rest of the systemic examination was normal. A CT scan of the brain did not show any abnormality.
His karyotype showed 54, XY, t(15; 22) (p 11; p 11). There was dicentric fusion
between chromosome 15 and chromosome 22. There were two centro meres present in the rearranged chromosome (Fig. 2). Both parents chromosomes were checked and they were normal.
At the age of 4 weeks he presented with vomiting and failure to gain weight. He was diagnosed as having gastro-esophageal reflux. At 13 months of age he was thought to be 3 to 4 months behind in development. His weight, length and head circumference continued to fall away from the 3rd centilc.
Fig. 1. The child aged 13 months-close up of the face.
Despite the family history this child did not have peroneal muscular atrophy type I on clinical grounds. Peroneal muscular atrophy has been localized to chromosome 17, but no mutation analysis or gene mapping was attempted in our patient.
Discussion
Though dicentric chromosomes resulting from the fusion of two acrocentrics have been described in live born children there do not appear to be any reports hitherto of such fusions between chromosomes 15 and 22. The dysmorphic features seen in our patient are relatively nonspecific and have been found to be associated with other cases of centric fusion, 15q deletion and in some children born to mothers treated with sodium valproate during pregnancy. The child reported by Fried- rich and co-workers(1) was a 15 months old boy with normal mental and physical development, his karyotype
showed a 15:22 balanced translocation. The other child reported by Mori and colleagues(2) was a two year old boy with dysmorphic features and psychomotor delay, karyotype showed a 15:22 translocation
which had resulted in partial monosomy of the long arm of chromosome 15.
There is a six to seven fold increase of malformations in the babies exposed to valproate compared to the general population(6). There is a tendency for the frequency of abnormalities and malformations to be related to the dose of valproate in the first trimester(7, 12). Maternal total valproate levels decrease as pregnancy progresses(l3). However, maternal free fractions of sodium valproate (expressed as per cent of total) have been shown to gradually increase during pregnancy although there was a wide variability in values particularly in late gestation(14). The sodium valproate free fraction are extremely high in fetal serum during early pregnancy
(40-80%
during week 13-26) but decrease to
approximately
20%
around week 20 of gestation and then gradually drop to 10% or below at end of gestation(l4).
Fig. 2.
Partial Karyotype
showing the dicentric fusion of chromosome No.
15 with No. 22.
Our child did not show any withdrawal symptoms which are said to be common, nine
of the 17 infants reported earlier(l2) showed manifestations of withdrawal. The
frequency of withdrawal symptoms is significantly related to the dose of valproate given to the mother in the thirdtrimester(12). Our infant also had hypoglycemia which has been reported(4,12).
Studies of the effects of sodium valproate have shown no evidence of an increase in chromosome breakages or any other chromosome
rearrangements(l5). It remains speculative as to whether exposure of the developing
oocyte could have caused the centre fusion in this case.
In general the risk of mental retardation or a physical malformation frama de novo
balanced translocatin is 0%-13% but for a de novo balanced Robertsonian translocation there is no discernible increased risk for phenotypic
abnormalities(l6). Indeed, as the fusion chromosome is dicentric in our patient, the implication is that there is no loss of long arm material from either chromosome. Therefore the combination of anomalies observed including facial characteristics, long philtrum, trigoncephaly and growth and motor retardation are due to the consequence of fetal expo- sure to sodium valptoate rather than the chromosome anomaly
per se.
Practitioners should therefore carry out reevaluation of clinical signs of patients where a chromosome anomaly is present in balanced form in a search of other associated causes as presented here.
|
1.
Friedrich U, Nielsen J, Sehested 1. A Family with 15-22 translocation. Hereditas 1972; 72: 172-174.
2.
Mori MA, Rodriguez L. Pinel I, Casas JM, Diaz De B, Martinez-Frias ML. Partial
monosomy 15q due to de ove t(15,22)(qI5; pi I). Ann Genetics 1987; 30: 246-248.
3.
Dalens B, Raynaud EJ, Gaulme J. Teratogenicity of valproic acid. J Pediatr 1980; 97: 332- 333.
4.
DiLiberi JH, Farndon P A, Dennis NR, Curry CJR. The fetal valproate syndrome. Am J Med Genet 1984; 19: 473-481.
5.
Winter RM, Donnai D, Bum J, Tucker SM. Fetal valproate syndrome: Is there a recognizable phenotype? J Med Genet 1987; 24: 692-695.
6.
Clayton-Smith J, Donnai D. Fetal valproate syndrome. J Med Genet 1995; 32: 724-727.
7.
Jager-Roman E, Deichi A, Jakob S. Fetal growth, major malformations, and minor anomalies in infants born to women receiving valproic acid. J Pediatr 1986; 108: 997-1004.
8.
Okada T, Tomoda T, Hisakawa H, Kurashige T. Fetal valproate syndrome with
reduction deformity of limb. Acta Paediatr Jpn 1995; 37: 58-60.
9.
Boussemart T, Bonneau D, Levard G, Berthier M, Oriot D. Omphalocele in a newborn baby exposed to sodium valproate in utero. Bur J Paediatr 1995; 154: 220-221.
10.
Williams PG, Hersh J H. A male with fetal valproate syndrome and autism. Dev Med Child Neuro11997; 39: 632-634.
11.
Christianson AL, Chesler N, Kromberg 10. Fetal valproate syndrome: Clinical and neuro-de velopmental features in two sibling pairs. Dev Med Child Neuro11994; 36: 361-369.
12.
Thisted E, Ebbesen F. Malformations, Withdrawal manifestations, and hypoglycemia after exposure to valproate in utero. Arch Dis Child 1993; 69: 288-291.
13.
Yerby MS, Friel PN, McCormick K. Antiepileptic drug disposition during preg-
nancy. Neurology 1992; 42 (Suppl.
5);
12-16.
14.
Nau H, Krauer B. Serum protein binding of valproic acid in fetus-mother pairs through- out pregnancy: Correlation with oxytocin administration and albumin and free fatty acid concentrations. J Clin Pharmacol 1986; 26: 215-221.
15.
Smith A, Beran RG. Chromosome fragility in the Lennox-Gastraut Epilepsy
Syndrome. J Intellect Disabil Res 1992; 36: 435-441.
16.
Weiss L, Van Dyke DL, Robertson J. The risk of mental retardation/multiple congenital anomalies related to de novo chromosome rearrangements. A first-order approximation. Pediatr Res 1983;
17: 221 A.
|