|
|
Case Reports Indian Pediatrics 2001; 38: 297-300 |
||||||||
|
Pleomorphic Xanthoastrocytoma |
||||||||
|
Pleomorphic xanthoastrocytoma (PXA) is a rare primary neoplasm of the brain. It was first described by Keeps et al.(1) in 1979 as a distinctive astrocytic neoplasm with a comparatively good prognosis. It has a characteristic appearance on neuroimaging. A case of a child with PXA is reported here as there are hardly any pediatric cases reported from India.
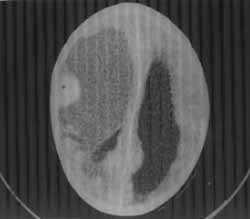
An 11-year-old right-handed boy presented with complaints of headache, vomiting, repeated episodes of seizures and progressive loss of vision for the last 10 months. The tonic-clonic seizures were of focal onset with secondary generalization. Each seizure lasted for 15-20 minutes and was followed by Todd’s paralysis and post-ictal drowsiness for one hour. In the past 5 months he had also developed asymmetry of the face and left sided hemiparesis. On examination, his vital parameters were stable. Macewan’s sign was positive indicating sutural separation. Except for dysarthric speech, his higher functions were normal. He had a searching nystagmus without perception of light. Fundus examination revealed bilateral optic atrophy. He had left sided supranuclear facial palsy and left sided hemiparesis. Bilaterally, deep tendon reflexes were brisk and the plantar responses were extensor. A CT scan was performed which showed a large hypodense well-delineated cystic lesion spanning the frontoparietotemporal region compressing the third ventricle and causing a midline shift with moderate dilation of both the lateral ventricles (Fig. 1). The cyst had an iso-dense mural nodule adherent to the meningeal aspect of the frontal lobe. It showed a uniform and brilliant enhancement on contrast. The cyst wall showed no enhancement. He underwent an operation wherein the tumor was completely excised. The brownish cystic mass contained a 1.5 × 0.8 × 0.8 cm adherent mural nodule. The cyst was filled with a brownish gelatinous substance. The nodule had a yellow homo-geneous appearance on the cut section. There were no areas of calcification or necrosis. Microscopically, the nodule consisted of round cells with vesicular nuclei, spindle-shaped proliferating cells with processes, a large number of xanthochromic cells and multi-nucleated giant cells with scattered lympho-cytes. There were no areas of necrosis or mitosis. Correlating the prolonged course that pointed to a slow growing tumor, the CT scan findings of a cystic mass with brilliantly enhancing mural nodule with meningeal attachement and histopathological findings, a diagnosis of pleomorphic xanthoastrocytoma was made. After surgery, the patient made an uneventful recovery. Although his neurological deficit improved, there was no change in his vision.
Pleomorphic xanthoastrocytoma is a recently described tumor that belongs to the well-circumscribed variety of astrocytic glial neoplasms(2). It is considered to have a neuroectodermal origin as the cytoplasm of tumor cells shows the presence of both glial fibrillary acid protein (GFAP) and S-100 protein(3). The peak age of onset is 20 years and 90% of the reported cases are below 30 years of age. Cases in the pediatric age group have been described. However, only one case in the pediatric age group has so far been reported in the Indian literature(4). PXA is a slow growing tumor and seizures constitute the initial manifestation. As the tumor grows, focal deficits and signs of raised intra-cranial pressure may appear(5,6). The tumor has classical neuroimaging characteristics. The CT scan, as in our case, reveals a hypodense cystic mass with distinct borders. An ecentric mural nodule attached to the meninges is seen. The nodule enhances uniformly and brilliantly on contrast. Mild edema may be present around the mass but calcifications are unusual. Usually, the cyst wall does not enhance. MRI shows a well-delineated cystic mass that appears hypo-or-iso-intense on T1-weighted images. The peripheral nodule enhances on contrast administration(7,8). Histopathologically, the pleomorphic tumor cells have multi-lobed nuclei, multi-nucleated giant cells, spindle cells and foamy lipid laden xanthomatous astrocytes are also seen(8,9). The tumor despite its pleomorphic appearance, has a low-grade malignant potential(8,10) and complete excision is usually curative. It does not require post-operative radiation therapy or chemo-therapy(11,12). Uncommonly though, the tumor may recur or demonstrate aggressive clinical behavior with a mortality rate between 15% and 20%.
|
![]()