|
|
|
Indian Pediatr 2021;58:650-666 |
 |
Consensus Guidelines on
Management of Steroid-Resistant Nephrotic Syndrome
|
|
Anil Vasudevan,1 Ranjeet
Thergaonkar,2 Mukta Mantan,3
Jyoti Sharma,4 Priyanka Khandelwal,5
Pankaj Hari,5 Aditi Sinha,5
Arvind Bagga,5 Expert Group
of Indian Society of Pediatric Nephrology*
From 1Department of Pediatric Nephrology, St. John’s Medical College
Hospital, Bengaluru; 2INHS Asvini, Mumbai; 3Maulana Azad Medical
College, New Delhi; 4Pediatric Nephrology Service, King Edward Memorial
Hospital, Pune; 5Division of Nephrology, Department of Pediatrics, All
India Institute of Medical Sciences, New Delhi, India.
*List of expert group members provided in Annexure I.
Correspondence to: Dr Arvind Bagga, Division of Nephrology,
Department of Pediatrics, All India Institute of Medical Sciences, New
Delhi 110 029, India.
Email: [email protected]
Published online: January 4, 2021;
PII:S097475591600278
|
Justification: The management of steroid
resistant nephrotic syndrome (SRNS) is challenging. These guidelines
update existing 2009 Indian Society of Pediatric Nephrology
recommendations on its management. Objective: To frame revised
guidelines on diagnosis and evaluation, treatment and follow up, and
supportive care of patients with the illness. Process: The
guidelines combine evidence-based recommendations and expert opinion.
Formulation of key questions was followed by systematic review of
literature, evaluation of evidence by experts and two face-to-face
meetings. Recommendations: Fourteen statements provide updated
advice for managing steroid resistance, and underscore the importance of
estimating proteinuria and baseline kidney function, and the need for
kidney biopsy and genetic screening. Calcineurin inhibitors are
recommended as most effective in inducing remission of proteinuria, the
chief factor associated with long-term renal survival. Advice on
managing allograft recurrence, congenital nephrotic syndrome, and
monitoring and supportive care, including transition of care, are
described. This revised practice guideline is intended to improve
management and patient outcomes, and provide direction for future
research.
Keywords: Calcineurin inhibitors, Congenital nephrotic
syndrome, Focal segmental glomerulosclerosis, Minimal change disease.
|
|
T
he prevalence of
idiopathic nephrotic
syndrome, characterized by proteinuria,
hypoalbuminemia and edema, varies from 12-16 per 100000 children [1]. Majority of patients achieve
remission of proteinuria following 4-6 weeks therapy with
prednisolone. However, 10-15% patients do not achieve complete
remission, and are termed steroid-resistant nephrotic synd-rome
(SRNS) [2]. Renal histology shows focal segmental
glomerulo-sclerosis (FSGS), minimal change disease and
mesangio-proliferative glomerulonephritis. Other patterns,
includ-ing C3 glomerulopathy, membranous nephropathy and IgA
nephropathy, and secondary causes of nephrotic syndrome are
uncommon. The management of patients with SRNS is challenging.
The illness is associated with unsatisfactory patient-reported
quality of life, morbidity due to infectious and non-infectious
illnesses, and side effects of therapy [2,3]. Patients with
persistent protein-uria are at risk for progressive kidney
failure [4].
Guidelines from the Indian Society of
Pediatric Nephrology (ISPN) were first published in 2009 [5]. In
view of recent evidence, the ISPN has proposed revision of these
recommendations. The revised guidelines refer to patients with
SRNS due to minimal change disease, mesangioproliferative
glomerulonephritis and FSGS. These guidelines also address
management of patients with post-transplant recurrence of FSGS
and congenital nephrotic syndrome. Clinical practice
recommendations, from the International Pediatric Nephrology
Association (IPNA), on the illness were published recently [6].
PROCESS
Three work-groups were constituted to
evaluate evidence on: (i) diagnosis and evaluation, (ii)
treatment and follow up, and (iii) supportive care of
patients with SRNS. The groups developed key questions, and
reviewed and analyzed published studies. Quality of evidence was
assessed and rated from A-D following the GRADE model [7], and
is provided with each guideline. Each statement was assigned one
of the two levels of guidance, recommen-dation or suggestion,
indicating strength of the advice (Web Table I).
Ungraded statements (X) are like practice points, not supported
by sufficient evidence.The work-groups discussed the evidence,
through alternating break-out and plenary sessions, in New Delhi
on 5 April 2019. Draft guidelines were discussed with members of
the ISPN in Pune on 21 December 2019.
GUIDELINES
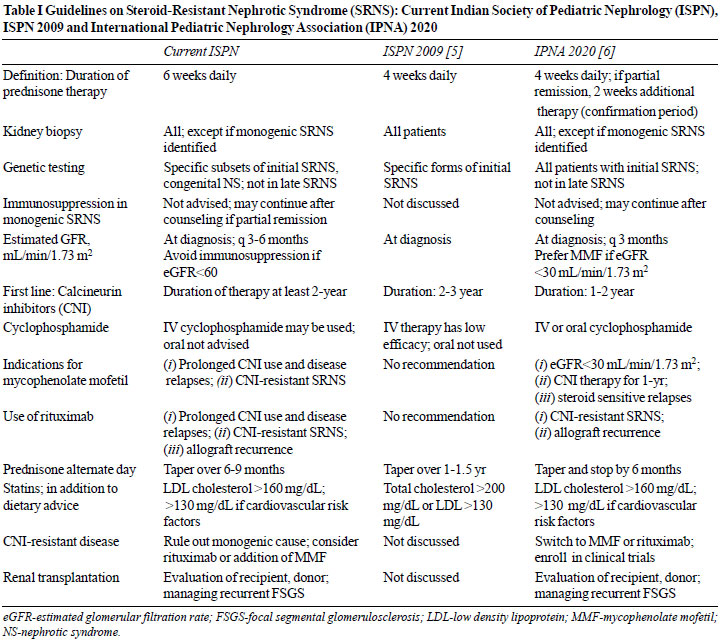
Table I compares
the current and previous guidelines [5] and recent
recommendations from the IPNA [6]. Given the challenges in
management, we advise that a pediatric nephrologist be
responsible for the diagnosis and management of children with
SRNS.
 |
| |
|
Box I Definitions Related to Nephrotic
Syndrome
Nephrotic syndrome
Nephrotic range proteinuria (40
mg/m2/h or > 1000 mg/m2/day; spot Up/Uc
³ 2
mg/mg; 3-4+ by dipstick); hypoalbuminemia (albumin
< 3.0 g/dL); and edema
Steroid sensitive nephrotic
syndrome
Complete remission within
6-weeks’ treatment with prednisolone at a dose of 60
mg/m2/day (2 mg/kg/day; maximum 60 mg/day)
Initial steroid-resistance
Failure to achieve complete
remission after 6-weeks initial therapy with
prednisolone (as defined above)
Late (secondary)
steroid-resistance
Initially steroid-sensitive;
steroid resistance in a subsequent relapse
Complete remission
Urine protein nil-trace by
dipstick for 3 consecutive days, Up/Uc < 0.2, or
24-h protein < 100 mg/m2/day
Partial remission
Urine protein 1+/2+ (dipstick),
Up/Uc between 0.2-2, or 24-h urine protein 100-1000
mg/m2/day; serum albumin
³ 3.0
g/dL; and absence of edema
Non-response
Urine protein 3+/4+ (dipstick),
Up/Uc ³ 2,
or 24-h urine protein > 1000 mg/m2/day; albumin
< 3.0 g/dL or edema
Relapse
Urine albumin 3+/4+ for 3
consecutive days, Up/Uc
³ 2,
or 24-h protein > 1000 mg/m2/day, in a patient
previously in partial or complete remission
Monogenic disease
Pathogenic or likely pathogenic
variation, defined by American College of Medical
Genetics and Genomics, in a gene associated with
steroid-resistant nephrotic syndrome (Web Table II)
CNI-resistant disease
Non-response to cyclosporine or
tacrolimus, given in adequate doses and titrated to
optimal blood trough levels, for 6-months
Allograft recurrence of nephrotic
syndrome
Persistent proteinuria (Up/Uc
> 1) if previously anuric; or increase of Up/Uc by
>1 if proteinuria at time of transplant (in absence
of other apparent causes)
CNI-calcineurin inhibitor; Up/Uc-urine protein to
creatinine ratio (mg/mg).
|
Guideline 1: Diagnosis of Steroid-Resistant
Nephrotic Syndrome (SRNS)
1.1 We recommend that
steroid-resistance be defined in patients not showing
complete remission of proteinuria, despite 6-weeks daily
treatment with prednisolone. (1B)
1.2 We suggest similar definitions for
initial and late (secondary) steroid-resistance (Box I).
(X)
Rationale
Approximately 85-90% patients with idiopathic
nephrotic syndrome respond to treatment with prednisolone, with
complete remission of proteinuria and normalization of serum
albumin [1]. There is lack of consensus regarding the minimum
duration of daily prednisolone treatment before defining
steroid-resistance. The International Study of Kidney Disease in
Children (ISKDC) reported that, of patients who achieved
remission, 94% did so within 4-weeks daily treatment and the
rest during 4-weeks’ alternate-day therapy [8]. Others found
that 4-weeks and 6-8 weeks initial therapy results in remission
in 90-92% and 87-93% patients, respectively [9-12]. While few
experts suggest additional therapy with 3-doses of IV methyl
pre-dnisolone before labeling steroid-resistance, this is not
uniformly practiced [6,13,14].
The previous version of this guideline
defined SRNS as lack of complete remission despite 4-weeks
therapy with prednisolone at a daily dose of 60 mg/m 2
[5]. The ISKDC and Kidney Disease: Improving Global Outcomes
(KDIGO) proposed that steroid-resistance be defined following
8-weeks therapy [8,15]. Recent IPNA and KDIGO guidelines propose
confirming steroid-resistance following 4-6-weeks’ therapy with
predniso(lo)ne, with or without additional therapy with
three-doses of IV methylprednisolone [6,16].
In order to balance the benefits of extending
therapy with steroid adverse effects, we recommend defining SRNS
in patients who fail to show complete remission of proteinuria
despite 6-weeks therapy with prednisolone at daily dose of 60
mg/m². Patients with steroid adverse effects may receive daily
prednisolone for 4-weeks, followed by alternate-day therapy for
the next 2-weeks. We do not advise therapy with IV
methylprednisolone before making the diagnosis of SRNS.
We suggest similar definitions for initial
(primary) and late (secondary) steroid-resistance (Box I). Initial
resistance is lack of remission at the first episode of
nephrotic syndrome. Patients who are steroid-sensitive initially
but show steroid-resistance during subsequent relapse have late
resistance. Systemic infections may be associated with
persistent proteinuria and should be treated appropriately.
Guideline 2: Evaluation of Patients
We recommend the following in all patients
with SRNS: Quantitation of proteinuria; serum creatinine;
estimated glomerular filtration rate (eGFR); and kidney biopsy (Box
II). (1A)
|
Box II Initial Evaluation of Patients
with Steroid-Resistant Nephrotic Syndrome
Urinalysis, including microscopy
Spot urine protein to creatinine
ratio; 24-h urine protein excretion
Complete blood counts
Blood creatinine, albumin,
electrolytes, fasting glucose, glycosylated hemoglobin
(HbA1c)
Total, low density and high-density
cholesterol; triglycerides
Calcium, phosphate, alkaline
phosphatase
Hepatitis B surface antigen;
hepatitis C and human immuno-deficiency virus antibodies
Ultrasonography of kidneys
Kidney biopsy (light,
immunofluorescence, electron micro-scopy); avoided in
selected patients*
Investigations in selected children
Complement C3, C4; antinuclear
antibody
Genetic tests: Initial
steroid-resistance with: (i) onset during
infancy; (ii) family history of
steroid-resistance, (iii) extrarenal features,
(iv) non-response to calcineurin inhibitors, (v)
prior to transplantation
Biopsy may be avoided in patients with familial
steroid-resistance or with extrarenal features, where
genetic diagnosis is preferred; a biopsy is also not
required in patients with congenital nephrotic syndrome
(Web Box II).
|
Rationale
Nephrotic syndrome is characterized by
nephrotic range proteinuria:
³ 3+ by
dipstick, proteinuria ³ 40
mg/m2/hr (> 1000
mg/m2/day), urine
protein to creatinine ratio (Up/Uc)
³ 2 mg/mg;
hypoalbuminemia (<3 g/dL); and edema [6]. All patients should be
evaluated appropriately (Box II). Estimation of
proteinuria, by Up/Uc in morning specimen or 24-hr protein
excretion, at diagnosis and 6-monthly follow-up, helps determine
response to therapy. Since 24-hr collection of urine is
difficult to implement, Up/Uc is preferred. Parents are
counseled regarding the importance of urinary dipstick analysis
for home monitoring of proteinuria.
Response of proteinuria to therapy is an
important determinant of renal survival [4,17,18]. Data from the
PodoNet Registry on 1354 patients with SRNS shows that 10-year
renal survival was highest (94%) in complete remission, 72% with
partial remission and 43% with non-response [19]. Assessment of
creatinine and eGFR at baseline and follow-up identifies acute
kidney injury (AKI) secondary to hypovolemia, fluid loss,
infections and drug toxicity, and CKD [20,21].
History and examination might help identify
genetic and secondary forms of SRNS. History of deafness,
developmental delay, seizures, family history of similar
disorder and consanguinity, and syndromic features or extrarenal
anomaly (e.g., genitourinary abnormality, microcoria, dystrophic
nails and microcephaly) suggest a genetic etiology. History of
joint pain, weight loss, alopecia, jaundice, rash or palpable
purpura indicates a secondary cause.
All patients with SRNS should undergo a
kidney biopsy before instituting specific treatment. Biopsies
are examined by light, immunofluorescence and electron
microscopy. An adequate biopsy should include the
corticomedullary junction and have ~20 glomeruli to identify
focal pathology like FSGS [22]. A biopsy is useful for: (i)
identifying pathology, extent of interstitial fibrosis and
glomerulosclerosis for diagnosis and prog-nosis; and (ii)
excluding differential diagnosis and secondary causes of
nephrotic syndrome. Repeat biopsy is required to assess
calcineurin inhibitor (CNI) toxicity, progression of disease or
change in pathology.
Chief histological diagnoses in children with
SRNS include FSGS (40-50%), minimal change disease (25-40%) and
mesangioproliferative glomerulonephritis (5-8%) [23]. Histology
suggestive of FSGS is considered a risk factor for progression
to CKD [15-17,24]. Around 10-15% patients show membranous
nephropathy, IgA nephropathy or proliferative
glomerulonephritis, which requires additional evaluation. A
kidney biopsy is not necessary in patients with well described
monogenic form of SRNS, known to be unresponsive to
immuno-suppression, e.g., congenital nephrotic syndrome,
familial disease, or if a known genetic cause is already
identified.
Screening for viral infections: Patients
should be evaluated for hepatitis B and C, and HIV infections.
Collapsing FSGS may be associated with HIV or parvovirus
infection [25]. Those with positive serology are evaluated for
viral load and extent of disease. Active infection may require
the use of antiviral therapy.
Guideline 3: Indications for Genetic Studies
We recommend genetic studies in the following
patients: congenital nephrotic syndrome; initial resistance
during infancy; nephrotic syndrome with extrarenal features;
familial steroid-resistance; non-response to therapy with CNI;
and prior to transplantation. (1B)
Rationale
Approximately 20-30% patients with SRNS have
pathogenic variations in genes encoding proteins of podocyte
structure and function (Web Table II) [2].
Mutations in NPHS1, NPHS2, WT1, COQ2, PLCE1 and LAMB2
account for 50-60% of monogenic disease in children [26-28].
Genetic testing is useful as follows:
• Identification of causal variant
enables diagnosis of monogenic disorders, and occasional
phenocopies (e.g., Alport syndrome, Dent disease,
cystinosis). Specific diagnosis allows counseling regarding
progression of kidney disease and monitoring for extrarenal
complications, e.g., patients with WT1,
LMX1B,WDR73 and SMARCAL1 mutations [29].
• Patients with monogenic etiology have
4-fold risk of non-response to therapy with CNI (odds ratio,
OR 4.00; 95% CI 2.52-6.51) and 3-fold risk of kidney failure
(OR 2.87; 95% CI 2.22-3.72)
(Web Table III)
[18,26,28,30].
• Certain mutations respond to targeted
therapy, e.g., coenzyme Q10 for defects in CoQ pathway, and
eplerenone for ARHGDIA mutations [31,32].
• Compared to patients with no
identifiable genetic cause, those with monogenic etiology
have significantly lower risk for allograft recurrence
[18,27,33].
• Diagnosis of a monogenic etiology
assists in counseling for future pregnancies and antenatal
diagnosis, and facilitates screening of live related renal
transplant donors [34-36].
While IPNA guidelines suggest comprehensive
genetic evaluation in all children with initial
steroid-resistance [6], we suggest a focused approach. The
likelihood of detecting a genetic cause is inversely related to
age at onset of the illness. A monogenic etiology was seen in
69%, 50%, 25%, 18% and 11% with disease presenting during the
first 3 months, 4-12 months, 1-6 years, 7-12 year and 13-18
years, respectively [26]. Syndromic forms of the illness may be
associated with specific mutations and characteristic phenotype
(Web Table II). Family history of similar illness or
consanguinity suggests a genetic cause in ~50-70% cases [26,27].
Although patients with an underlying genetic etiology are less
likely to respond to therapy with CNI, few patients may
occasionally show partial remission [37].
Siblings of patients with a monogenic cause
may be screened for proteinuria by dipstick. There is no role
for genetic screening in healthy children with family history of
the disease. Since pathogenic mutations are not identified in
patients with late steroid-resistance, genetic testing in these
children is also not indicated [18,27].
The precise prevalence of monogenic
variations in Indian patients with SRNS is unclear as studies
are limited to small cohorts [38,39]. A nationwide study is in
progress to determine the genetic basis of SRNS, and indications
for testing may be revised in future.
Method of Genetic Testing
Causal variants in ~90 genes are associated
with monogenic SRNS (Web Table II). Most genes do
not show a clear phenotype-genotype correlation. Next-generation
sequencing (NGS) panels, incorporating multiple genes relevant
to the phenotype, are feasible and less expensive, and provide
higher diagnostic yield than Sanger sequen-cing. These panels
include genes associated with other renal diseases that may have
phenotype similar to SRNS. Clinical exome sequencing (Mendeliome
gene panel), which includes all exons of genes listed in Online
Mendelian Inheritance of Man (OMIM) database, facili-tates
targeted gene analysis. In case a causative variant is not
identified in the gene-panel, search for variants may be
extended to remaining genes in the clinical exome. Whole exome
sequencing might be considered for novel disease-causing genes.
Sanger sequencing is preferred if a disease-causing mutation is
highly likely in a specific gene, in context of extrarenal
features or positive family history with known genetic cause.
Sanger sequencing is essential to confirm variants detected on
NGS, to screen parents to confirm segregation and for antenatal
counseling.
Parents should be advised regarding risks and
benefits of NGS, including limitation of insurance cover.
Referral to genetic counselors might be necessary. Testing must
be performed by certified and experienced laboratories, and
pathogenicity of variants determined based on criteria proposed
by the American College of Medical Genetics and Genomics [40].
Guideline 4: Therapy of Patients with SRNS
4.1 We recommend calcineurin inhibitors
(CNI) as first-line therapy for patients with initial or
late steroid-resistance. (1A)
4.2 We suggest continuing therapy with
CNI for at least 24-months if partial or complete remission
is achieved. (2C)
4.3 We suggest that CNI therapy
should be withheld or discontinued for patients with AKI
stage 2-3 or estimated glomerular function rate (eGFR)
persistently below 60 ml/min/1.73m 2.
(2C)
Rationale
Therapy aims to induce complete or partial
remission, while avoiding medication-related toxicity. Long-term
renal outcome in patients who achieve remission is significantly
better when compared to non-responders [17-19,41]. Randomized
controlled trials (RCT) and case series show that therapy with
CNI (cyclosporine, tacrolimus) results in complete remission in
30-40% and complete or partial remission in 60-80% patients
[2,3,18,41,42]. A Cochrane meta-analysis that compared
cyclosporine to no treatment showed increased likelihood of
complete or partial remission with the former (2 RCT; relative
risk RR 3.50; 95% CI 1.04-9.57) at 6-months [43]. Similarly,
therapy with CNI, compared to IV cyclophosphamide, was
associated with higher rates of complete or partial remission (3
RCT; RR 1.98; 95% CI 1.25-3.13) [43]. While most reports do not
show different outcomes between initial and late
steroid-resistance [44-46], better outcomes in the latter have
been reported [18]. The efficacy of tacrolimus and cyclosporin
is comparable (2 RCT; RR 1.05; 95% CI 0.87-1.25), with no
difference in nephrotoxicity or hypertension [43,47].
Similar to the IPNA and KDIGO guidelines, we
recommend first-line use of CNI for patients with SRNS [6,16].
Tacrolimus is preferred to cyclosporine except in children who
are unable to swallow tablets (cyclosporine is available as
suspension), and patients with seizures or at risk for diabetes.
Doses of tacrolimus and cyclosporine are titrated to achieve
recommended trough levels, keeping in mind interaction with
other medications (Table II and
Web Table IV).
Low levels are associated with non-response and relapse, while
high levels increase the risk for nephrotoxicity [48]. Lower
levels may be targeted once sustained remission is achieved for
6-9 months [49,50]. Fig. 1 provides an outline of the
approach to management of SRNS.
 |
| |
 |
|
Fig. 1 Management of
steroid-resistant nephrotic syndrome. Kidney biopsy is
necessary, except in patients where genetic testing may
obviate the need for biopsy (Box II). Patients with
monogenic cause for steroid-resistance should not
receive immunosuppression and are managed with
angiotensin converting enzyme (ACE) inhibitors and
supportive therapy. Patients with likely non-genetic
disease are initiated on therapy with a calcineurin
inhibitor (CNI) along with supportive care. Lack of
remission despite adequate therapy with CNI for 6-months
is an indication for genetic screening, if not performed
earlier. Patients with CNI-resistant disease who do not
show a monogenic defect may be treated with IV rituximab
or combined therapy of CNI and mycophenolate mofetil
(MMF). Immunosuppression is withdrawn in patients with
continued non-response.
|
Most patients who respond to CNI do so within
the first 6-months of treatment [44,45,47,51]. Non-response to
CNI is therefore considered in patients who continue to show
nephrotic-range proteinuria, hypoalbuminemia or edema despite
6-months therapy. Patients showing non-response should be
screened for significant genetic variations (see above), and
considered for alternate management (Guideline 6).
Therapy with CNI is initially combined with
prednisolone, administered at a dose of 1-1.5 mg/kg on alternate
days for 4-6 weeks, and tapered over 6-9 months [6,44-46].
Following CNI-induced remission, ~60% patients may have
steroid-sensitive relapses [44,45,52]. Relapses are treated with
prednisolone (2 mg/kg/day until remission; tapered on
alternate-days). Stoppage of steroid therapy might not be
possible in patients with multiple relapses.
The duration of treatment with CNI for
patients with partial or complete remission is not clear, with
guidelines recommending minimum 12-months’ therapy [6,16]. An
RCT comparing continued therapy with tacrolimus vs switching to
mycophenolate mofetil (MMF) at 6-months, found the former twice
as effective in maintaining remission (90% vs 45%) [45].
In a retrospective study on 23 patients, therapy with
cyclosporine for mean duration of 1.7 years could be
successfully switched to MMF in 79% cases [52]. In view of the
risk of relapse with early cessation of therapy, we suggest
continuing therapy with CNI for 24 months or longer (Fig. 1),
ensuring adequate dose and trough levels [49,51].
About 10-25% patients receiving prolonged CNI
treatment are at risk of nephrotoxicity [53]. Risk factors for
nephrotoxicity include presence of initial resistance, dose of
CNI used, duration of heavy proteinuria, and hyper-tension
during therapy [48,53]. In order to balance the benefits and
toxicity of CNI, we suggest individualizing therapy in children
with partial or complete response at 24-months. Options include:
i) discontinue therapy if patient has been in sustained
remission; ii) continue CNI therapy; perform kidney
biopsy if treatment is prolonged beyond 30-36 months, or if
restarting treatment; iii) switch to IV rituximab or oral
MMF in patients with CNI or steroid toxicity or
steroid-sensitive relapses.
Risk factors for AKI in nephrotic syndrome
include volume depletion, infections, nephrotoxic injury and
steroid resistance [21,54,55]. We suggest withholding CNI during
AKI [16,55,56]; treatment is restarted following recovery of
kidney function. Therapy with CNI is avoided if eGFR is
persistently <60 mL/min/1.73 m 2.
Guideline 5: Alternate Immunosuppressive
Therapy
5.1 We suggest treatment with IV
cyclophosphamide in patients with non-availability of CNI,
either due to its cost or adverse effects. (2B)
5.2 We do not suggest the use of
oral cyclophosphamide for therapy of patients with
steroid-resistance. (2A)
Rationale
Studies utilizing IV cyclophosphamide (every
month for 6-months) and tapering prednisolone show complete or
partial remission in 10-50%, but with significant adverse
effects [46,57,58]. Compared to CNI, IV cyclo-phosphamide is
associated with lower rates of sustained remission (RR 0.50; 95%
CI 0.37-0.68) at 6-months [43]. A multicenter study compared the
efficacy of cyclosporine (150 mg/m 2/day)
for 48-weeks with IV cyclophosphamide (500 mg/m2;
7-doses over 36 weeks) in patients with SRNS. While complete
remission was low, 47% patients treated with cyclosporine and 6%
with IV cyclophos-phamide had partial response [57]. Another
multicenter trial on 131 patients showed 6-month complete
remission rates of 14.8% and partial remission rates of 31.1%
with IV cyclophosphamide, as against 52.4% and 30.1%,
respectively with tacrolimus [44].
Two RCT showed similar efficacy and safety of
oral and IV cyclophosphamide in 61 children with
steroid-resistance (RR 1.58; 95% CI 0.65-3.85) [58,59]. However,
two other RCT found no difference in rates of remission in
patients receiving oral cyclophosphamide with predni-sone
compared to prednisone (n=84; RR 1.06, 95% CI 0.61-1.87)
[60,61]. Based on the above, we do not advise use of oral
cyclophosphamide in patients with SRNS.
Guideline 6: Treatment of CNI-Resistant
Nephrotic Syndrome
In patients with non-genetic forms of SRNS
and non-response to therapy with CNI, we suggest additional
treatment with either IV rituximab or oral MMF (Fig. 1).
(2C)
Rationale
Approximately 25-35% patients with
non-genetic forms of SRNS do not show complete or partial
remission following 6-months’ therapy with CNI [43]. The
management of patients with non-response to CNI therapy is
difficult, since they are at high risk of kidney failure
[17-19]. Patients with initial steroid- and CNI-resistance
should be screened for an underlying monogenic disorder. Those
with no pathogenic or likely pathogenic variants in podocyte
genes may be considered for additional immuno-suppressive
therapy, administered under close supervision.
While rituximab has shown promising results
in patients with steroid-sensitive nephrotic syndrome, its
efficacy in CNI-resistant SRNS is less satisfactory. In a
systematic review (7 case series, one RCT; n=226) on
efficacy of rituximab in steroid and CNI-resistant nephrotic
syndrome, the mean number of rituximab doses was 3.1. Complete
or partial remission was observed in 46.4%, with better response
in minimal change disease (63.2%) than in FSGS (39.2%), and
late-resistance (52.8%) compared to initial-resistance (40.8%)
[62]. Similar findings of satisfactory response to rituximab in
patients with late resistance are reported in a series from
United Kingdom [18] and in a systematic review [63]. While less
favorable outcomes were reported in a study from India, with
remission in 29.3% of 58 patients with CNI-resistance, there was
trend for better response in minimal change disease and
late-resistance [64].
We suggest administering 2-doses of IV
rituximab at a dose of 375 mg/m 2
at weekly interval, targeting CD19 count <5/µl or
£1% of
lymphocyte count. If CD19 target is not met, 1-2 additional
doses may be repeated at weekly intervals (maximum 4 doses). In
patients achieving complete or partial remission, repeat dose(s)
of rituximab may be given following B-cell reconstitution, which
typically occurs after 6-9 months. There is limited guidance
regarding redosing with rituximab, and benefits should be
balanced by the risk of side effects, including infusion
reactions, serum sickness, neutropenia and
hypo-gammaglobulinemia. Therapy with rituximab may be associated
with reactivation of hepatitis B, Pneumocystis jirovecii
pneumonia, severe lung injury and rarely, progressive multifocal
leukoencephalopathy [65].
The efficacy of MMF in patients with SRNS is
less satisfactory than in steroid-sensitive disease. In the
PODONET cohort, monotherapy with this medication was not
effective in 83% patients [19]. The efficacy of combination of
CNI and MMF (600 to 1000 mg/m2/day)
has been reported in patients with CNI-resistant disease. Three
case-series (n=168) on combined therapy for 6-12 months,
show complete remission, partial remission and non-response in
11.8-47.7%, 8.7-38.2% and 43.5-58.8%, respectively [66-68].
There is limited data on the efficacy of treatment with
adalimumab, abatacept, ofatumumab and adrenocorticotrophic
hormone, oral galactose and LDL apheresis in patients with
CNI-resistant SRNS. These therapies should only be used in
context of clinical trials [69-71].
Intense immunosuppression is associated with
risk of systemic infections. Patients receiving combined therapy
with CNI and either rituximab or MMF should receive prophylaxis
with cotrimoxazole (5 mg/kg trimethoprim on alternate days) for
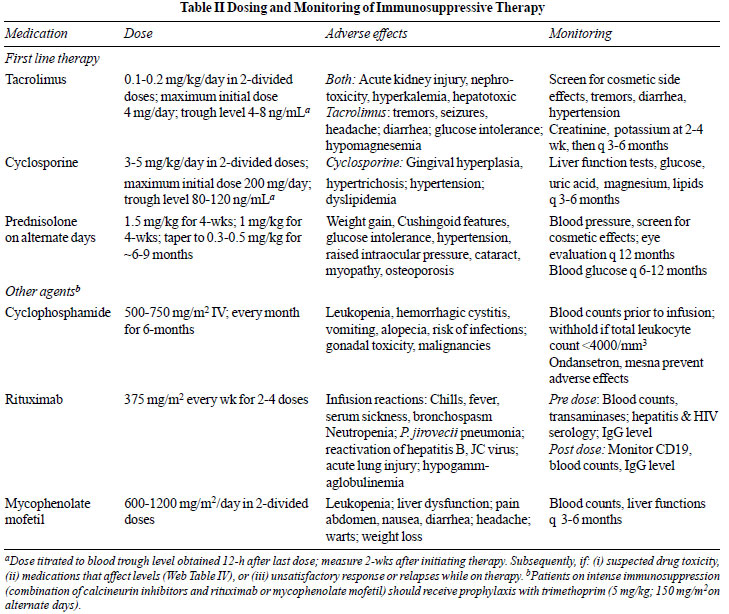
3-6 months. Table II summarizes dosing, side effects and
monitoring of children receiving immunosuppressive agents.
Guideline 7: Immunosuppressive Therapy With
Pathogenic or Likely Pathogenic Variants
We do not recommend that patients with
monogenic disease receive therapy with calcineurin inhibitors or
other immunosuppressive agents. (1B)
Rationale
Patients with SRNS with pathogenic or likely
pathogenic variations (monogenic disease, Box I) usually
do not show complete or partial remission following therapy with
CNI. Analysis of pooled data (Web Table III; n=867)
shows that compared to non-genetic disease, those with genetic
forms of SRNS are not likely to respond to CNI (RR 4.00; 95% CI
2.52-6.51). Patients with monogenic forms of SRNS, irrespective
of response are more likely to progress to kidney failure than
those with non-genetic illness (RR 2.87; 95% CI 2.22-3.72).
The recent IPNA guidelines do not recommend
that patients with monogenic disease receive immuno-suppressive
medications [6]. However, some patients with a genetic cause for
steroid-resistance, especially those with WT1 variants,
might show partial remission following treatment with CNI [37].
The decision to continue therapy in such patients should follow
counseling of parents regarding anticipated benefits (relief of
edema, higher blood albumin) vs risks (therapy-related
toxicity, infec-tions) and cost of therapy. Targeted therapy is
possible for specific mutations, e.g., coenzyme Q10 for
defect(s) in CoQ10 pathway, eplerenone for ARHGDIA, and
cortico-steroids for mutations in genes of Rho/Rac/Cdc42 network
[31,32].
Guideline 8: Angiotensin Converting Enzyme
Inhibitors and Angiotensin Receptor Blockers
We recommend that all patients with SRNS
should receive therapy with angiotensin converting enzyme (ACE)
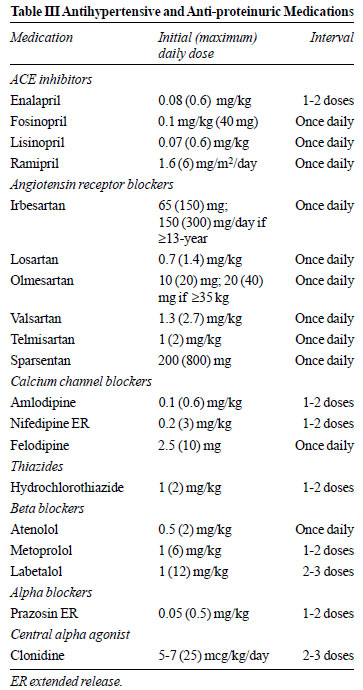
inhibitors or angiotensin receptor blockers (ARB) (Table III).
(1B)
Rationale
Since proteinuria is a risk factor for
progressive kidney disease, its reduction is important for
renoprotection [72]. Use of ACE inhibitors is associated with
30-40% reduction in proteinuria in a dose- and time-dependent
manner (16,43). ARB may be used as effectively (Table III)
[73]. Dual blockade with ACE inhibitors and ARB further
reduces proteinuria, but is associated with side effects such as
hypotension, AKI and hyperkalemia, and is not recommended [74].
ACE inhibitors or ARB are avoided in patients with eGFR < 25
mL/min/1.73 m 2, and
discontinued during vomiting, diarrhea or reduced oral intake.
In patients with FSGS, sparsentan, that combines endothelin
receptor type A blockade with angiotensin II inhibition, reduces
proteinuria and hypertension more effectively than irbesartan
[75]. We do not advise therapy with other medications that
target the renin-angiotensin axis, including aliskrein,
eplerenone and vitamin D analogs.
SUPPORTIVE CARE AND MONITORING
Important aspects of supportive care are
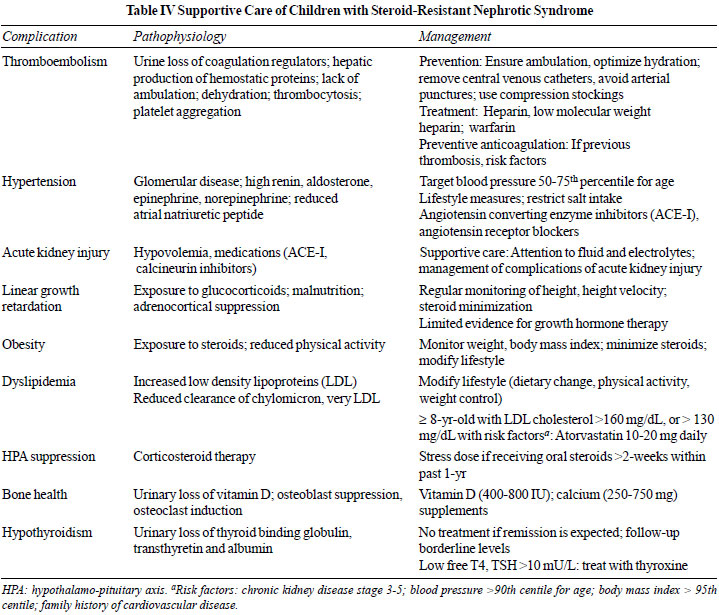
summarized in Table IV. Principles of management of
edema, systemic infections and immunization are discussed in the
revised ISPN guidelines on steroid-sensitive nephrotic syndrome,
published recently [76].
Guideline 9: Thrombotic Complications
We do not recommend routine
thromboprophylaxis in children with SRNS. (1C)
Rationale
The risk of thromboembolic complications in
nephrotic syndrome is ~3% in children, compared to 25% in
adults, with most events within the first 3-months of illness
[77]. Risk factors for thrombosis include congenital nephrotic
syndrome, heavy proteinuria, membranous nephropathy, central
venous catheters and coexisting heart disease [77]. Sites of
thrombosis include the deep veins, cerebral sinus(es), renal
veins and occasionally, arteries [78].
Routine use of prophylactic anticoagulants is
not recommended [77]. Aspirin is less effective and is
associated with risk of AKI [79]. Non-pharmacological measures
such as ambulation, hydration and use of compression stockings
are encouraged; central venous catheters and arterial punctures
should be avoided [79,80].
Therapy aims to prevent extension of thrombi
and reduce the risk of embolism. Thrombolysis followed by
anticoagulation is considered in patients with life or
limb-threatening thrombosis. While anticoagulation may be
initiated with unfractionated heparin, this requires IV access
and close laboratory monitoring, has less pre-dictable
pharmacokinetics and is associated with the risk of adverse
effects (thrombocytopenia, anaphylaxis and osteoporosis) [80].
Use of low-molecular weight heparin is preferred [79,81].
Therapy is initiated with enoxaparin at a dose of 1.5 mg/kg/dose
(<2-months age) or 1 mg/kg/dose (>2-months) subcutaneously,
every 12-hr [81]. Long-term therapy may continue either with
enoxaparin or warfarin (0.2 mg/kg/dose started concurrently with
enoxaparin) for 3-months or until remission [80]. For warfarin
the international normalized ratio (INR) for prothrombin time is
targeted between 2.0 and 3.0. Children with recurrent
throm-botic events require long-term anticoagulation [77,80].
Guideline 10: Cardiovascular Morbidity
We recommend strategies to minimize
cardiovascular risk in patients with SRNS (X).
Rationale
Steroid resistance is associated with
multiple cardio-vascular risks, including hypertension,
dyslipidemia, hypoalbuminemia, hypercoagulable state and
steroid-induced obesity. Strategies to reduce this risk include
minimizing residual proteinuria, managing hypertension, weight
reduction to achieve BMI <85th centile for age, non-exposure to
tobacco, and achieving target levels of lipids, fasting glucose
(<100 mg/dL) and HbA1c (< 5.7%) [82].
Hypertension: Blood pressure should be
measured at each visit. A study on Indian children with
frequently relapsing disease showed clinic hypertension in 64%,
ambulatory hypertension in 33%, white coat hypertension in 30%
and increased left ventricular mass in 21% [83]. Systolic and
diastolic blood pressures are targeted between 50-75th
percentile for age and sex [84]. Lifestyle changes include
increased intake of vegetables, fresh fruits, low-fat milk,
legumes and nuts, and reduced salt and sweets. Pharmacotherapy
is initiated with ACE inhibitor or ARB, in view of additional
benefit of reducing proteinuria (Table III).
Dyslipidemia: Children with nephrotic
syndrome show high blood levels of cholesterol, triglycerides,
apoB-containing lipoproteins (LDL, VLDL, IDL) and lipoprotein (a).
While abnormalities resolve during remission, these might
persist in patients with SRNS. Dyslipidemia aggravates
glomerulosclerosis and proximal tubular damage and is associated
with progression of CKD. Screening for dyslipidemia is advised
in patients with SRNS, and those with steroid-sensitive disease
and cardiovascular risk factors [82,85].
We advise reduced intake of trans-fats or
saturated fats and sugar, and increased consumption of fruits,
vegetables, legumes and whole grain cereals [85]. The CHILD-1
diet is the first step in children with dyslipidemia or risk
factors for cardiovascular disease and includes restricting
intake of saturated fat and cholesterol to <10% of daily
calories and 300 mg, respectively. In case this is not
effective, the respective restrictions are enhanced to 7% and
200 mg in the CHILD-2 diet [82,85]. Limiting leisure screen time
to <2-hr/day, ensuring moderate physical activity for 1-hr/day,
and vigorous physical activity at least 3 days a week are
advised [85].
If lifestyle measures fail to correct
dyslipidemia, therapy with statins is advised, especially if
associated with risk factors for cardiovascular disease [85].
Therapy in children 8-year or older may begin with atorvastatin
at 10 mg/day, with monitoring for adverse effects.
Guideline 11: Stress Dosing of
Glucocorticoids
We recommend that patients, who have received
oral corticosteroids for more than 2-weeks within the past
one-year, should receive additional steroid dosing during
conditions associated with physiological stress. (1D)
Rationale
Therapy for nephrotic syndrome involves
high-dose prednisolone for 12-weeks for the first episode, 5-6
weeks for relapse, and prolonged alternate-day for frequent
relapses and steroid-resistance. A systematic review reported
that 269 of 487 (55.2%) children receiving corticosteroids for
varied indications for more than 14-days had biochemical
evidence of suppressed hypothalamo-pituitary axis (HPA) [86].
The duration of HPA suppression might last up to two years, and
vary with dose and duration of treatment [87].
We recommend additional steroids in
situations where physiological stress is expected (fever
³38°C,
inadequate oral intake, lethargy, dehydration, invasive surgery,
dental surgery, trauma and large burns). Conditions such as
uncomplicated viral infections, acute otitis media and fever
post-immunization do not require stress dosing. In case of
critical illness or surgery, hydrocortisone is administered
parenterally at 100 mg/m2,
initially or preoperatively followed by 25 mg/m2
every 6-hr. With less serious illness, hydrocortisone 30-50 mg/m2/day
or prednisolone 0.3-1.0 mg/kg in a single daily dose is given
during stress and tapered thereafter [88].
Guideline 12: Monitoring of Patients
Children with SRNS are at risk for
progression to stage 5 CKD, complications of the disease and
adverse effects of medications [89-91]. Managing
immunosuppressive therapies is a challenge due to the risk of
infections, non-compliance and presence of co-morbidities.
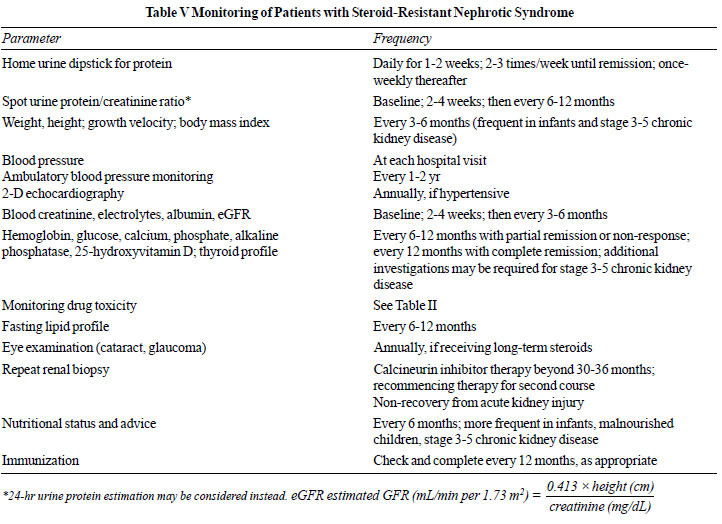
Patients require regular monitoring and careful follow up, and
counseling regarding need for compliance with medications
(Table V).
Guideline 13: Transplantation
13.1 We recommend that kidney
transplant be considered in all patients with SRNS and stage
5 CKD. (1B)
13.2 We recommend that genetic
testing be performed before transplant to assist in donor
selection and predict the risk of recurrence in allograft.
(1B)
13.3 In a patient with prior
allograft recurrence, the decision for retransplantation
should be taken after discussing the risks and benefits with
treating physicians, patient and family. (2C)
13.4 In patients with allograft
recurrence, we suggest initiation of plasma exchanges,
increasing the dose of CNI, with or without additional use
of rituximab. (2B)
Rationale
Kidney transplantation is the definitive
option for patients with SRNS and stage 5 CKD. Careful
pre-transplant evaluation of recipient and donor is required.
Genetic screening of the recipient is necessary, particularly if
there is initial resistance or equivocal course of the illness,
since it stratifies the risk for allograft recurrence and helps
in donor screening. If inheritance pattern is autosomal
recessive, a heterozygous carrier (parent) may be accepted as a
donor with negligible risk of recurrence, except Afro-Caribbean
donors with APOL1 risk variant, or heterozygous R229Q
variants in NPHS2 [35,92]. Heterozygous carriers
of pathogenic variants in COL4A3 and COL4A4 and
women with variants in COL4A5 should not be accepted as
donors since they are at risk of kidney failure [93]. For
autosomal dominant inheritance, individuals with same variant
are not accepted as donors since they might show variable
penetrance with late onset of disease.
FSGS recurs in the allograft in ~30% (range
6-50%) patients [94,95]. Recurrence is associated with allograft
dysfunction and its loss in 40-60% patients, especially in those
with persistent nephrotic range proteinuria [33,96]. Recurrence
risk is highest in patients with late steroid resistance or
recurrent nephrotic syndrome in a prior transplant (~80%),
moderate with initial resistance and no identified genetic cause
(~50%), and lowest with confirmed genetic mutation underlying
SRNS (<5%) [18,97-100]. Patients with FSGS and kidney failure
should be counseled about these risks.
Living-related transplantation is associated
with better graft survival and is preferred for children in our
country. While the risk of recurrence is minimally higher in
children receiving live-related grafts, this is balanced by
reduced risk of rejection and lower need for immunosuppression
[100,101]. Live-related trans-plantation is therefore the first
choice, except in patients with moderate to high risk of
recurrence.
Nephrotic syndrome might recur occur within
hours to days after transplant and is characterized by nephrotic
range proteinuria and progressive hypoalbuminemia. Patients are
monitored for recurrence by screening for proteinuria (Up/Uc
ratio), initially daily and then with reduced frequency (Web
Box I). Recurrence is considered in patients with
proteinuria and Up/Uc ³1
mg/mg if anuric prior to transplant or increase of ratio by
³1 in
those with proteinuria at transplantation [6]. Early onset graft
dysfunction may be a feature of recurrent FSGS. Where feasible,
an allograft biopsy is recommended to detect podocyte foot
process effacement or segmental sclerosis that supports the
diagnosis of recurrence. A biopsy may also help exclude other
diagnosis in patients with lower degree of late-onset
proteinuria or allograft dysfunction.
Multiple therapies have been used to prevent
recurrence of nephrotic syndrome, including pre-transplant
plasma exchanges, rituximab and lipoprotein apheresis. There is
limited evidence that any of these strategies prevent allograft
recurrence in the first kidney transplant [102,103]. Strategies
for managing patients with allograft recurrence include
combination of plasma exchanges with high-dose CNI and
corticosteroids, with or without cyclophosphamide [104-107]
(Web Box I). Multiple reports show benefit from
additional therapy with rituximab (2-4 doses of 375 mg/m2,
administered once every 1-2 weeks) [65,104]. Using these
strategies, 60-70% patients with recurrent FSGS show complete or
partial remission.
Guideline 14: Transition of Care
A significant proportion of patients continue
to have active disease into adulthood [89]. These children will
need to be cared for by ‘adult’ physicians and nephrologists,
keeping with the policy of the Indian Academy of Pediatrics of
caring for children upto 18 years [108]. Parallel to the change
in medical caregiver, patients need to transition from care by
parents to self-care. Transition should occur smoothly, without
affecting patient health. Institution-specific protocols for
transition of care should be based on standard guidelines [109].
Congenital Nephrotic Syndrome
Patients with congenital nephrotic syndrome
present at birth or in first 3-months of life. Infants are born
prematurely with large placenta, and show massive proteinuria,
hypoalbuminemia and anasarca. Antenatal ultrasonography may show
hyperechoic kidneys; amniocentesis reveals high
alpha-fetoprotein. There may be dysmorphic features or
comorbidities. Most patients develop kidney failure by the age
of 2-8 years. Recommendations on genetic aspects and management
were published recently [110,111].
Almost 70-80% patients with congenital
nephrotic syndrome have a genetic cause; mutations in NPHS1,
NPHS2, WT1, LAMB2 and PLCE1 account for ~90% cases
[110,112]. Exome sequencing using an extended SRNS gene panel
(Web Table II) is recommended. Results of screening
have implications for genetic counseling. Rarely, the condition
is secondary to intrauterine infec-tions with cytomegalovirus,
rubella, toxoplasma and syphilis [111]. The role of kidney
biopsy is limited and may be considered if a genetic diagnosis
is not established.
Evaluation aims to confirm the diagnosis and
identify complications, including poor growth, hypothyroidism,
systemic infections and thromboembolism (Web Box II)
[111]. Infants with WT1 variants are monitored by
ultrasonography for Wilms tumor every 3-6 months.
Management includes maintaining euvolemia,
optimizing nutrition, and therapy of complications. Patients
should receive high energy (110-120 Cal/kg) and protein (3-3.5
g/kg/d) diet, orally or by feeding gastrostomy. Supplements of
thyroxine, vitamin D and calcium are required. Albumin infusions
(0.5-1.0 g/kg) are advised in presence of hypovolemia (oliguria,
prolonged capillary refill, tachycardia) or anasarca. IV
furosemide (0.5-2 mg/kg) is given at the end of infusion, unless
patient has features of hypovolemia. Monitoring of fluid status,
creatinine, electrolytes and blood pressure are necessary during
diuretic therapy [111].
After 4-weeks of life, judicious use of ACE
inhibitors (Table III) with or without prostaglandin
inhibitors (indomethacin, celecoxib) is effective in reducing
the severity of proteinuria. Therapy with these agents and
diuretics should be withheld during episodes of hypovolemia.
Since infections are the chief cause of death, infants should
receive all primary immunization and bacterial infections are
treated promptly. Therapy with anticoagulants is considered in
patients with history of thrombosis.
Unilateral or bilateral nephrectomies are not
proposed routinely, and may be considered in patients with
repeated episodes of hypovolemia or refractory edema, thrombosis
and malnutrition [112]. Bilateral nephrectomy is advised, prior
to kidney transplantation, in patients with WT1 mutations
or persistent nephrotic range proteinuria. Kidney
transplantation is the definitive treatment, but has ethical,
technical and immunologic challenges.
CONCLUSIONS
Recommendations on management of SRNS, first
proposed by the ISPN in 2009, have been revised based on
systematic reviews, published studies and expert opinion. While
there is better understanding regarding the genetic basis and
management, important clinical issues require to be examined
(Box III). The management of the disease continues to be
challenging, and patients not responsive to treatment with CNI
are at risk of progressive kidney disease. We hope that the
present guidelines will standardize therapies and improve the
quality of care for these patients.
|
Box III Research Priorities in
Steroid-Resistant Nephrotic Syndrome
Determine genetic burden and
genotype-phenotype correlation in Indian patients;
models for evaluating functional significance of
variants
Pathogenesis of non-genetic forms of
the illness
Duration of therapy with calcineurin
inhibitors; switching to less toxic medications
Treatment for patients who are
non-responsive to therapy with calcineurin inhibitors
Prevention and therapy for recurrent
focal segmental glomerulosclerosis
Improving quality of life and patient-centered
outcomes.
|
Note: Supplementary material related to
this study is available with the online version at
www.indianpediatrics.net
Contributors: All authors involved in
review of literature and preparation of background document; AV,
RT, MM, JS, AS and AB drafted the manuscript; AB conceived the
idea and critically revised the manuscript. All authors approved
the final version of the manuscript.
Funding: Indian Council of Medical
Research; Advanced Centre for Research in Pediatric Kidney
Diseases; 5/7/1090/2013-RHN; Department of Biotechnology,
Government of India; BT/PR11030/MED/30/1644/2016.
Competing interests: None stated.
ANNEXURE I
*List of Participants
Kamran Afzal, Aligarh; Indira Agarwal,
Vellore; Vinay Agarwal, New Delhi; Kanav Anand,
New Delhi; M Ashraf, Srinagar; Arvind Bagga, New
Delhi; Sushmita Banerjee, Kolkata; Girish C Bhatt,
Bhopal; Sudha Ekambaram, Chennai; Arpita Gogoi,
Dibrugarh; Sanjeev Gulati, New Delhi; Pankaj Hari,
New Delhi; Suprita Kalra, New Delhi; Kanika Kapoor,
New Delhi; Priyanka Khandelwal, New Delhi; Sriram
Krishnamurthy, Puducherry; Manish Kumar, New Delhi;
Mukta Mantan, New Delhi; Jitendra K Meena, New Delhi;
Kirtisudha Mishra, New Delhi; Amarjeet Mehta, Jaipur;
OP Mishra, Varanasi; Aliza Mittal, Jodhpur; Saroj
K Patnaik, New Delhi; Subal Pradhan, Cuttack; PK
Pruthi, New Delhi; Sumantra Raut, Kolkata;
Abhijeet Saha, New Delhi; Manisha Sahay, Hyderabad;
Jyoti Sharma, Pune; Shobha Sharma; New Delhi;
Jyoti Singhal, Pune; Aditi Sinha, New Delhi; Rajiv
Sinha, Kolkata; Ranjeet Thergaonkar, Mumbai;
Karalanglin Tiewsoh, Chandigarh; Susan Uthup,
Thiruvananthapuram; Anand S Vasudev, New Delhi; Anil
Vasudevan, Bengaluru.
Experts: Uma Ali, Mumbai; Amit K Dinda, New
Delhi; Mohammed Faruq, New Delhi; Madhuri Kanitkar,
New Delhi; Kumud Mehta, Mumbai; BR Nammalwar,
Chennai; Kishore D Phadke, Bengaluru; Geetika Singh,
New Delhi; RN Srivastava, New Delhi.
REFERENCES
1. Noone DG, Iijima K,
Parekh R. Idiopathic nephrotic syndrome in children. Lancet.
2018;392:61-74.
2. Tullus K, Webb H,
Bagga A. Management of steroid-resistant nephrotic syndrome
in children and adolescents. Lancet Child Adolesc Health.
2018;2:880-90.
3. Gipson DS, Trachtman
H, Kaskel FJ, et al. Clinical trial of focal segmental
glomerulosclerosis in children and young adults. Kidney Int.
2011;80:868-78.
4. Troyanov S, Wall CA,
Miller JA, Scholey JW, Cattran DC. Focal and segmental
glomerulosclerosis: Definition and relevance of a partial
remission. J Am Soc Nephrol. 2005;16:1061-8.
5. Indian Society of
Pediatric Nephrology, Gulati A, Bagga A, Gulati S, Mehta KP,
Vijayakumar M. Management of steroid resistant nephrotic
syndrome. Indian Pediatr. 2009;46:35-47.
6. Trautmann A,
Vivarelli M, Samuel S, et al. IPNA clinical practice
recommendations for the diagnosis and management of children
with steroid-resistant nephrotic syndrome. Pediatr Nephrol.
2020;35:1529-61.
7. American Academy of
Pediatrics Steering Committee on Quality Improvement and
Management. Classifying Recommendations for Clinical
Practice Guidelines. Pediatrics. 2004;114: 874-7.
8. The primary nephrotic
syndrome in children. Identification of patients with
minimal change nephrotic syndrome from initial response to
prednisone: A report of the International Study of Kidney
Disease in Children. J Pediatr. 1981;98:561-4.
9. Sinha A, Saha A,
Kumar M, et al. Extending initial prednisolone
treatment in a randomized control trial from 3 to 6 months
did not significantly influence the course of illness in
children with steroid-sensitive nephrotic syndrome. Kidney
Int. 2015;87:217-24.
10. Nakanishi K, Iijima
K, Ishikura K, et al. Two-year outcome of the ISKDC
regimen and frequent-relapsing risk in children with
idiopathic nephrotic syndrome. Clin J Am SocNephrol.
2013;8:756-62.
11. Bagga A, Hari P,
Srivastava RN. Prolonged versus standard prednisolone
therapy for initial episode of nephrotic syndrome. Pediatr
Nephrol. 1999;13:824-7.
12. Hoyer PF, Brodeh J.
Initial treatment of idiopathic nephrotic syndrome in
children: Prednisone versus prednisone plus cyclosporine A:
A prospective, randomized trial. J Am Soc Nephrol. 2006;
17:1151-7.
13. Murnaghan K, Vasmant
D, Bensman A. Pulse methylprednisolone therapy in severe
idiopathic childhood nephrotic syndrome. Acta Paediatr
Scand. 1984;73:733-9.
14. Letavernier B,
Letavernier E, Leroy S, Baudet-Bonneville V, Bensman A,
Ulinski T. Prediction of high-degree steroid dependency in
pediatric idiopathic nephrotic syndrome. Pediatr Nephrol.
2008;23:2221-6.
15. Kidney Disease:
Improving Global Outcomes (KDIGO) Glomerulonephritis Work
Group. KDIGO Clinical Practice Guideline for
Glomerulonephritis. Kidney Int Suppl. 2012;2:139-274.
16. Rovin BH, Caster DJ,
Cattran DC, et al; Conference Participants. Management and
treatment of glomerular diseases (part 2): Conclusions from
a Kidney Disease: Improving Global Outcomes (KDIGO)
Controversies Conference. Kidney Int. 2019; 95:281-95.
17. Gipson DS, Chin H,
Presler TP, et al. Differential risk of remission and ESRD
in childhood FSGS. Pediatr Nephrol. 2006;21:344-9.
18. Mason AE, Sen ES,
Bierzynska A, et al. Response to first course of
intensified immunosuppression in genetically stratified
steroid resistant nephrotic syndrome. Clin J Am Soc Nephrol.
2020;15: 983-94.
19. Trautmann A,
Schnaidt S, Lipska-Ziêtkiewicz BS, et al. Long-term
outcome of steroid-resistant nephrotic syndrome in children.
J Am Soc Nephrol. 2017;28:3055-65.
20. Stevens PE, Levin A;
Kidney Disease: Improving Global Outcomes (KDIGO) Chronic
Kidney Disease Guideline Development Work Group Members.
Evaluation and management of chronic kidney disease:
Synopsis of the Kidney Disease: Improving Global Outcomes
2012 Practice Guideline. Ann Intern Med. 2013;158: 825-30.
21. Kushwah S, Yadav M,
Hari P, Meena J, Sinha A, Bagga A. Incidence and
determinants of acute kidney injury in patients with
nephrotic syndrome. Asian J Pediatr Nephrol. 2019; 2:75-81.
22. Corwin HL, Schwartz
MM, Lewis EJ. The importance of sample size in the
interpretation of the renal biopsy. Am J Nephrol.
1988;8:85-9.
23. Gulati S, Sengupta
D, Sharma RK, et al. Steroid resistant nephrotic syndrome:
Role of histopathology. Indian Pediatr. 2006;43:6.
24. D’Agati VD, Fogo AB,
Bruijn JA, Jennette JC. Pathologic classification of focal
segmental glomerulosclerosis: A working proposal. Am J
Kidney Dis. 2004;43:368-82.
25. Wenderfer SE.
Viral-associated glomerulopathies in children. Pediatr
Nephrol. 2015;30:1929-38.
26. Sadowski CE, Lovric
S, Ashraf S, et al. A single-gene cause in 29.5% of
cases of steroid-resistant nephrotic syndrome. J Am Soc
Nephrol. 2015;26:1279-89.
27. Bierzynska A,
McCarthy HJ, Soderquest K, et al. Genomic and
clinical profiling of a national nephrotic syndrome cohort
advocates a precision medicine approach to disease
management. Kidney Int. 2017;91:937-47.
28. Trautmann A,
Lipska-Ziêtkiewicz BS, Schaefer F. Exploring the clinical
and genetic spectrum of steroid resistant nephrotic
syndrome: The PodoNet Registry. Front Pediatr. 2018;6:200.
29. Landini S, Mazzinghi
B, Becherucci F, et al. Reverse phenotyping after
whole-exome sequencing in steroid-resistant nephrotic
syndrome. Clin J Am Soc Nephrol. 2020;15:89-100.
30. Nagano C, Yamamura
T, Horinouchi T, et al. Comprehensive genetic diagnosis of
Japanese patients with severe proteinuria. Sci Rep.
2020;10:270
31. Montini G,
Malaventura C, Salviati L. Early coenzyme Q10
supplementation in primary coenzyme Q10 deficiency. N Engl J
Med. 2008;358:2849-50.
32. Gee HY, Saisawat P,
Ashraf S, et al. ARHGDIA mutations cause nephrotic syndrome
via defective RhoGTPase signaling. J Clin Invest.
2013;123:3243-53.
33. Morello W,
Puvinathan S, Puccio G, et al. Post-transplant
recurrence of steroid resistant nephrotic syndrome in
children: The Italian experience. J Nephrol. 2020;33:849-57.
34. Hildebrandt F,
Heeringa SF. Specific podocin mutations determine age of
onset of nephrotic syndrome all the way into adult life.
Kidney Int. 2009;75:669-71.
35. Lentine KL, Kasiske
BL, Levey AS, et al. KDIGO Clinical Practice
Guideline on the evaluation and care of living kidney
donors. Transplantation. 2017;101:S1-S109.
36. Lipska BS,
Iatropoulos P, Maranta R, et al. Genetic screening in
adolescents with steroid-resistant nephrotic syndrome.
Kidney Int. 2013;84:206-13.
37. Malakasioti G, Iancu
D, Tullus K. Calcineurin inhibitors in nephrotic syndrome
secondary to podocyte gene mutations: A systematic review.
Pediatr Nephrol. 2021;36:1353-64.
38. Siji A, Karthik KN,
Pardeshi VC, Hari PS, Vasudevan A. Targeted gene panel for
genetic testing of south Indian children with steroid
resistant nephrotic syndrome. BMC Med Genet. 2018;19:200.
39. Ramanathan ASK,
Vijayan M, Rajagopal S, Rajendiran P, Senguttuvan P. WT1 and
NPHS2 gene mutation analysis and clinical management of
steroid-resistant nephrotic syndrome. Mol Cell Biochem.
2017;426:177-81.
40. Richards S, Aziz N,
Bale S, et al. Standards and Guidelines for the
Interpretation of Sequence Variants: A Joint Consensus
Recommendation of the American College of Medical Genetics
and Genomics and the Association for Molecular Pathology.
Genet Med. 2015;17:405-24.
41. Troost JP, Trachtman
H, Nachman PH, et al. An outcomes-based definition of
proteinuria remission in focal segmental glomerulosclerosis.
Clin J Am Soc Nephrol. 2018;13:414-21.
42. Hamasaki Y,
Yoshikawa N, Nakazato H, et al. Prospective 5-year
follow-up of cyclosporine treatment in children with
steroid-resistant nephrosis. Pediatr Nephrol.
2013;28:765-71.
43. Liu ID, Willis NS,
Craig JC, Hodson EM. Interventions for idiopathic
steroid-resistant nephrotic syndrome in children. Cochrane
Database Syst Rev. 2019;11:CD003594.
44. Gulati A, Sinha A,
Gupta A, et al. Treatment with tacrolimus and prednisolone
is preferable to intravenous cyclophosphamide as the initial
therapy for children with steroid-resistant nephrotic
syndrome. Kidney Int. 2012;82:1130-5.
45. Sinha A, Gupta A,
Kalaivani M, Hari P, Dinda AK, Bagga A. Mycophenolate
mofetil is inferior to tacrolimus in sustaining remission in
children with idiopathic steroid-resistant nephrotic
syndrome. Kidney Int. 2017;92:248-57.
46. Mantan M, Sriram CS,
Hari P, Dinda A, Bagga A. Efficacy of intravenous pulse
cyclophosphamide treatment versus combination of intravenous
dexamethasone and oral cyclophosphamide treatment in
steroid-resistant nephrotic syndrome. Pediatr Nephrol.
2008;23:1495-502.
47. Choudhry S, Bagga A,
Hari P, Sharma S, Kalaivani M, Dinda A. Efficacy and safety
of tacrolimus versus cyclosporine in children with
steroid-resistant nephrotic syndrome: A randomized
controlled trial. Am J Kidney Dis. 2009;53:760-9.
48. Kim JH, Park SJ,
Yoon SJ, et al. Predictive factors for ciclosporin-associated
nephrotoxicity in children with minimal change nephrotic
syndrome. J Clin Pathol. 2011;64:516-9.
49. Inaba A, Hamasaki Y,
Ishikura K, et al. Long-term outcome of idiopathic
steroid-resistant nephrotic syndrome in children. Pediatr
Nephrol. 2016; 31:425-34.
50. Jahan A, Prabha R,
Chaturvedi S, Mathew B, Fleming D, Agarwal I. Clinical
efficacy and pharmacokinetics of tacrolimus in children with
steroid-resistant nephrotic syndrome. Pediatr Nephrol.
2015;30:1961-7.
51. Büscher AK, Beck BB,
Melk A, et al. Rapid response to cyclosporin A and favorable
renal outcome in nongenetic versus genetic steroid-resistant
nephrotic syndrome. Clin J Am Soc Nephrol. 2016;11: 245-53.
52. Gellermann J, Ehrich
JHH, Querfeld U. Sequential maintenance therapy with
cyclosporin A and mycophenolate mofetil for sustained
remission of childhood steroid-resistant nephrotic syndrome.
Nephrol Dial Transplant. 2012;27:1970-8.
53. Sinha A, Sharma A,
Mehta A, et al. Calcineurin inhibitor induced nephrotoxicity
in steroid resistant nephrotic syndrome. Indian J Nephrol.
2013; 23:41-6.
54. Sharma M, Mahanta A,
Barman AK, Mahanta PJ. Acute kidney injury in children with
nephrotic syndrome: A single-center study. Clin Kidney J.
2018;11:655-8.
55. Rheault MN, Zhang L,
Selewski DT, et al. AKI in children hospitalized with
nephrotic syndrome. Clin J Am Soc Nephrol. 2015;10: 2110-8.
56. Lombel RM, Gipson
DS, Hodson EM; Kidney Disease: Improving Global Outcomes.
Treatment of steroid-sensitive nephrotic syndrome: New
guidelines from KDIGO. Pediatr Nephrol. 2013;28: 415-26.
57. Plank C, Kalb V,
Hinkes B, et al. Cyclosporin A is superior to
cyclophosphamide in children with steroid-resistant
nephrotic syndrome: A randomized controlled multicentre
trial by the Arbeitsge-meinschaft für Pädiatrische
Nephrologie. Pediatr Nephrol. 2008;23: 1483-93.
58. Elhence R, Gulati S,
Kher V, Gupta A, Sharma RK. Intravenous pulse
cyclophosphamide - A new regime for steroid-resistant
minimal change nephrotic syndrome. Pediatr Nephrol. 1994;8:
1-3.
59. Shah KM, Ohri AJ,
Ali US. A randomized controlled trial of intravenous versus
oral cyclophosphamide in steroid-resistant nephrotic
syndrome in children. Indian J Nephrol. 2017;27:430-4.
60. International Study
of Kidney Disease in children. Prospective, controlled trial
of cyclophosphamide therapy in children with nephrotic
syndrome. Report of the International study of Kidney
Disease in children. Lancet. 1974;2:423-7.
61. Tarshish P, Tobin
JN, Bernstein J, Edelmann CM. Cyclophosphamide does not
benefit patients with focal segmental glomerulosclerosis. A
report of the International Study of Kidney Disease in
Children. Pediatr Nephrol. 1996;10:590-3.
62. Jellouli M, Charfi
R, Maalej B, Mahfoud A, Trabelsi S, Gargah T. Rituximab in
the management of pediatric steroid-resistant nephrotic
syndrome: A systematic review. J Pediatr. 2018;197:191-7.e1.
63. Kamei K, Ishikura K,
Sako M, Ito S, Nozu K, Iijima K. Rituximab therapy for
refractory steroid-resistant nephrotic syndrome in children.
Pediatr Nephrol. 2020;35:17–24.
64. Sinha A, Bhatia D,
Gulati A, et al. Efficacy and safety of rituximab in
children with difficult-to-treat nephrotic syndrome. Nephrol
Dial Transplant. 2015;30:96-106.
65. Sinha A, Bagga A.
Rituximab therapy in nephrotic syndrome: Implications for
patients’ management. Nat Rev Nephrol. 2013;9:154-69.
66. Wu B, Mao J, Shen H,
et al. Triple immunosuppressive therapy in
steroid-resistant nephrotic syndrome children with
tacrolimus resistance or tacrolimus sensitivity but
frequently relapsing. Nephrol (Carlton). 2015;20:18-24.
67. Okada M, Sugimoto K,
Yagi K, Yanagida H, Tabata N, Takemura T. Mycophenolate
mofetil therapy for children with intractable nephrotic
syndrome. Pediatr Int. 2007;49:933-7.
68. Nikibakhsh AA,
Mahmoodzadeh H, Karamyyar M, Hejazi S, Noroozi M, Macooie
AA. Treatment of steroid and cyclosporine-resistant
idiopathic nephrotic syndrome in children. Int J Nephrol.
2011;2011:930965.
69. Lee JM, Kronbichler
A, Shin JI, Oh J. Current understandings in treating
children with steroid-resistant nephrotic syndrome. Pediatr
Nephrol. 2021;36:747-61.
70. Muso E, Mune M,
Hirano T, et al. A prospective observational survey on the
long-term effect of LDL apheresis on drug-resistant
nephrotic syndrome. Nephron Extra. 2015;5:58-66.
71. Yu C-C, Fornoni A,
Weins A, et al. Abatacept in B7-1-positive proteinuric
kidney disease. N Engl J Med. 2014;370:1263-6.
72. van den Belt SM,
Heerspink HJL, Gracchi V, de Zeeuw D, Wühl E, Schaefer F.
Early proteinuria lowering by angiotensin-converting enzyme
inhibition predicts renal survival in children with CKD. J
Am Soc Nephrol. 2018;29:2225-33.
73. Webb NJA, Shahinfar
S, Wells TG, et al. Losartan and enalapril are comparable in
reducing proteinuria in children. Kidney Int.
2012;82:819-26.
74. Stotter BR, Ferguson
MA. Should ACE inhibitors and ARBs be used in combination in
children? Pediatr Nephrol. 2019;34: 1521-32.
75. Trachtman H, Nelson
P, Adler S, et al. DUET: A phase 2 study evaluating the
efficacy and safety of sparsentan in patients with FSGS. J
Am Soc Nephrol. 2018;29:2745-54.
76. Sinha A, Bagga A,
Banerjee S, et al; Expert group of Indian Society of
Pediatric Nephrology. Steroid Sensitive Nephrotic Syndrome:
Revised Guidelines. Indian Pediatr. 2021;58:461-81.
77. Kerlin BA, Ayoob R,
Smoyer WE. Epidemiology and pathophysiology of nephrotic
syndrome-associated thromboembolic disease. Clin J Am Soc
Nephrol. 2012;7:513-20.
78. Suri D, Ahluwalia J,
Saxena AK, et al. Thromboembolic complications in childhood
nephrotic syndrome: A clinical profile. Clin Exp Nephrol.
2014;18:803-13.
79. Kerlin BA, Haworth
K, Smoyer WE. Venous thromboembolism in pediatric nephrotic
syndrome. Pediatr Nephrol. 2014;29:989-97.
80. Monagle P, Chan AKC,
Goldenberg NA, et al. Antithrombotic Therapy in Neonates and
Children: Antithrombotic Therapy and Prevention of
Thrombosis, 9th edn. American College of Chest Physicians
Evidence - Based Clinical Practice Guidelines. Chest.
2012;141:e737S-801S.
81. Dabbous MK, Sakr FR,
Malaeb DN. Anticoagulant therapy in pediatrics. J Basic Clin
Pharm. 2014;5:27-33.
82. Hari P, Khandelwal
P, Smoyer WE. Dyslipidemia and cardiovascular health in
childhood nephrotic syndrome. Pediatr Nephrol.
2020;35:1601-19
83. Sarkar S, Sinha A,
Lakshmy R, et al. Ambulatory blood pressure monitoring in
frequently relapsing nephrotic syndrome. Indian J Pediatr.
2017;84:31-5.
84. Lurbe E,
Agabiti-Rosei E, Cruickshank JK, et al. 2016 European
Society of Hypertension Guidelines for the Management of
High Blood Pressure in Children and Adolescents. J
Hypertens. 2016;34:1887-920.
85. Expert Panel on
Integrated Guidelines for Cardiovascular Health and Risk
Reduction in Children and Adolescents, National Heart, Lung,
and Blood Institute. Expert panel on integrated guidelines
for cardiovascular health and risk reduction in children and
adolescents: Summary report. Pediatrics. 2011;128:S213-56.
86. Aljebab F, Choonara
I, Conroy S. Systematic review of the toxicity of
short-course oral corticosteroids in children. Arch Dis
Child. 2016;101:365-70.
87. Ahmet A, Mokashi A,
Goldbloom EB, et al. Adrenal suppression from
glucocorticoids: Preventing an iatrogenic cause of morbidity
and mortality in children. BMJ Paediatr Open. 2019;
3:e000569.
88. Liu D, Ahmet A, Ward
L, et al. A practical guide to the monitoring and management
of the complications of systemic corticosteroid therapy.
Allergy Asthma ClinImmunol. 2013;9:30.
89. Kaku Y, Ohtsuka Y,
Komatsu Y, et al. Clinical practice guideline for pediatric
idiopathic nephrotic syndrome 2013: General therapy. Clin
Exp Nephrol. 2015;19:34-53.
90. Foster BJ, Shults J,
Zemel BS, Leonard MB. Risk factors for glucocorticoid-induced
obesity in children with steroid-sensitive nephrotic
syndrome. Pediatr Nephrol.2006;21:973-80.
91. Simmonds J, Grundy
N, Trompeter R, Tullus K. Long-term steroid treatment and
growth: A study in steroid-dependent nephrotic syndrome.
Arch Dis Child. 2010;95:146-9.
92. Bierzynska A, Saleem
MA. Deriving and understanding the risk of post-transplant
recurrence of nephrotic syndrome in the light of current
molecular and genetic advances. Pediatr Nephrol. 2018;33:
2027-35.
93. Gross O, Weber M,
Fries JWU, Müller G-A. Living donor kidney transplantation
from relatives with mild urinary abnormalities in Alport
syndrome: Long-term risk, benefit and outcome. Nephrol Dial
Transplant. 2009;24:1626-30.
94. Francis A, Didsbury
M, McCarthy H, Kara T. Treatment of recurrent focal
segmental glomerulosclerosis post-kidney transplantation in
Australian and New Zealand children: A retrospective cohort
study. Pediatr Transplant. 2018;22: e13185.
95. Kienzl-Wagner K,
Waldegger S, Schneeberger S. Disease recurrence: The sword
of Damocles in kidney transplantation for primary focal
segmental glomerulosclerosis. Front Immunol. 2019;10:1669.
96. Vinai M, Waber P,
Seikaly MG. Recurrence of focal segmental glomerulosclerosis
in renal allograft: An in-depth review. Pediatr Transplant.
2010;14:314-25.
97. Dall’Amico R,
Ghiggeri G, Carraro M, et al. Prediction and treatment of
recurrent focal segmental glomerulosclerosis after renal
transplantation in children. Am J Kidney Dis.
1999;34:1048-55.
98. Uffing A, Pérez-Sáez
MJ, Mazzali M, et al. Recurrence of FSGS after kidney
transplantation in adults. Clin J Am Soc Nephrol. 2020;
15:247-56.
99. Ding WY, Koziell A,
McCarthy HJ, et al. Initial steroid sensitivity in children
with steroid-resistant nephrotic syndrome predicts
post-transplant recurrence. J Am SocNephrol. 2014;25:1342-8.
100. Koh LJ, Martz K,
Blydt-Hansen TD; NAPRTCS Registry Investigators. Risk
factors associated with allograft failure in pediatric
kidney transplant recipients with focal segmental
glomerulosclerosis. Pediatr Transplant. 2019;23:e13469.
101. Nehus EJ, Goebel
JW, Succop PS, Abraham EC. Focal segmental
glomerulosclerosis in children: Multivariate analysis
indicates that donor type does not alter recurrence risk.
Transplantation. 2013;96: 550-4.
102. Verghese PS,
Rheault MN, Jackson S, Matas AJ, Chinnakotla S, Chavers B.
The effect of peri-transplant plasmapheresis in the
prevention of recurrent FSGS. Pediatr Transplant. 2018;22:
e13154.
103. Shenoy M, Lennon R,
Plant N, Wallace D, Kaur A. Pre-emptive rituximab and plasma
exchange does not prevent disease recurrence following
living donor renal transplantation in high-risk idiopathic
SRNS. Pediatr Nephrol. 2020;35:1081-4.
104.Hansrivijit P,
Ghahramani N. Combined rituximab and plasmapheresis or
plasma exchange for focal segmental glomerulosclerosis in
adult kidney transplant recipients: A meta-analysis. Int
Urol Nephrol.2020;52:1377-87.
105.Kashgary A, Sontrop
JM, Li L, et al. The role of plasma exchange in treating
post-transplant focal segmental glomerulosclerosis: A
systematic review and meta-analysis of 77 case-reports and
case-series. BMC Nephrol. 2016;17:104.
106.Allard L, Kwon T,
Krid S, et al. Treatment by immunoadsorption for recurrent
focal segmental glomerulosclerosis after pediatric kidney
transplantation: A multicentre French cohort. Nephrol Dial
Transplant. 2018;33:954-63.
107. Cormican S, Kennedy
C, O’Kelly P, et al. Renal transplant outcomes in primary
FSGS compared with other recipients and risk factors for
recurrence: A national review of the Irish Transplant
Registry. Clin Transplant. 2018;32:e13152.
108. John TJ. IAP policy
on age of children for pediatric care. Indian
Pediatr.1999;36:461-3.
109. Watson AR, Harden
P, Ferris M, Kerr PG, Mahan J, Ramzy MF. Transition from
pediatric to adult renal services: A consensus statement by
the International Society of Nephrology and the
International Pediatric Nephrology Association. Pediatr
Nephrol. 2011;26:1753-7.
110.Lipska-Ziêtkiewicz
BS, Ozaltin F, Hölttä T, et al. Genetic aspects of
congenital nephrotic syndrome: A consensus statement from
the ERKNet–ESPN inherited glomerulopathy working group. Eur
J Hum Genet. 2020;28:1368-78
111.Boyer, O, Schaefer,
F, Haffner D, et al. Management of congenital nephrotic
syndrome: Consensus recommendations by the ERKNet-ESPN
Working Group. Nat Rev Nephrol.2021;17:277-89.
112.Joshi A, Sinha A, Sharma A, et al; NephQuest Consortium.
Next-generation sequencing for congenital nephrotic syndrome: A
multi-center cross-sectional study from India. Indian Pediatr.
2021;58: 445-51.
|
|
|
 |
|