|
|
Case Reports Indian Pediatrics 2001; 38: 93-96 |
|||||||||||||
|
Progressive Pseudorheumatoid Arthritis of Childhood |
|||||||||||||
|
Progressive pseudorheumatoid arthritis of childhood (PPAC) is a rare atuosomal recessive disorder, resembling rheumatoid arthritis, especially the seronegative spondylo-arthropathies of childhood(1). However, there is no inflammation of the joints and it has been demonstrated that the disorder is actually due to a non inflammatory chondropathy affecting mainly the articular cartilage(1,2). An early clinical diagnosis is difficult and it is the characteristic radiological features which help in an early recognition of the disease. Three cases have been reported from India earlier(3,5). We report here a case of PPAC presenting to us at the age of 11 years, who was diagnosed for the first time at our center on the basis of clinical and radiological features. However, this patient was different from the other reported cases in having prominent facial dysmorphism.
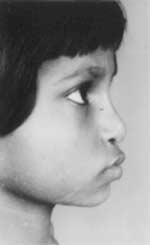
An 11-year-old girl, was brought with the complaints of progressively worsening stiffness and restriction of movements of all joints, associated with increasing difficulty in walking, and inability to close hands into a fist. She was the product of a non-consanguineous marriage; her mother was 24 years and father 30 years old. She was the second of three sibs, all of whom were normal. There was no history of a similar illness in any family member. She was born by a full term normal vaginal delivery. The antenatal, intranatal and neonatal periods were uneventful. She developed normally both physically and mentally till the age of 3 years, when she was noted to walk with her right leg and foot turned outwards. Within a year she developed knock knees as well. She also had complaints of being tired easily following exertion, difficulty in getting up from the squatting position and climbing stairs. The difficulty appeared to be related to the developing abnormality of the joints as well as to muscular weakness. At age five years she developed increasing stiffness of her joints which progressed to a marked restriction of movements of many of her joints namely elbows, wrists, small joints of hands and feet, hip, knee, ankle and neck at the time of presentation. There was no history of fever, joint pains or fractures. She had been worked up elsewhere, and been given provisional diagnoses of rheuma-toid arthritis, rickets, mucopolysaccharidosis and metaphyseal dysplasia; treatments received were high doses of Vitamin D, anti-inflammatory drugs and steroids at different points of time, without any significant relief. On examination, she was thin built, height was 120 centimetres falling below the 5th centile for her age, upper segment was 60 centimeres (upper segment = lower segment) and arm span-125 centimaetres (more than height). Vitals were within normal limits. She had a dysmorphic face with a mild depression of the nasal bridge, flattened nose, everted nostrils, hypoplastic maxilla and a prominent pouting mouth, giving a leonine appearance (Fig. 1). There were no corneal opacities.
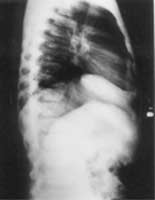
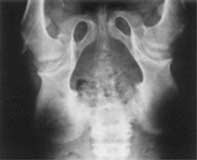
Examination of the musculoskeletal system examination showed widening of the elbows, wrists, knees and interphalangeal joints of the hands and feet. Movements of all joints were restricted, including neck and spine (with loss of lumbar lordosis), shoulder, elbow, wrist, knee, ankle and small joints of hands and feet, the restriction being maximum in the distal joints as compared to the proximal joints. The widening and fixed flexion deformity of the metacarpophalangeal and interphalangeal joints of the hands gave the hands a claw like appearance. There was also coxa-vara and genu valgum. There were no bony deformities or pain or tenderness of any of the joints. Gait was waddling and because of stiffness of all joints, like a puppet. Examination of central nervous system showed normal higher functions with IQ 90. Cranial nerves were normal. There was wasting of all muscles of hands and feet; tone and power could not be tested properly in view of the stiffness of all the joints. The deep tendon jerks were normal. Other neurological parameters were normal. Abdominal examina-tion did not reveal any hepatosplenomegaly, and the cardiovascular and respiratory systems were unreamarkable. Investigations revealed: Serum calcium 10.2 mg/dl, phosphorus 3.2 mg/dl, and alkaline phosphatase 12.0 KA units. Urine for muco-polysaccharidosis was negative. CPK was 115 units/1, and rheumatoid factor and antinuclear antibodies were negative. ESR was 9 mm fall in first hour by Westergren method. On skeletal survey, X-ray Chest and skull were normal. X-ray thoracolumbar spine (anteroposterior and lateral view) showed reduced height of the lumbar vertebral bodies. There was anterior erosion of the vertebral body, with a tongue like projection of the vertebra anteriorly. The intervertebral disc space was normal (Fig. 2). The femoral epiphysis appeared broad, with widened symphysis pubis on pelvic X-ray (Fig.3). There was metaphyscal and epiphyseal widening with irregularity of the epiphyseal margins on hand X-rays.
.
PPAC is an autosomal recessive dis-order(6); the gene has been localised recently to chromosome 6q22(7,8). The etiopatho-genesis is not clear, but it has been shown to be primarily a disorder of the articular cartilage. Histological studies show a peculiar nest like clustering of the chondrocytes in the resting and growth cartilage(1). Various treatment modalities have been tried, but none have proven effective till date. The disease is relentlessly progressive and leads to severe difficulties in ambulation and activities of daily living. No reports are available on the ultimate outcome of these patients since this is a relatively new disease. Slowly progressive restriction of move-ments of all the joints starting between 3-8 years age, associated with widening of the joints but no pain or tenderness, should raise the suspicion of PPAC. However, it is an uncommon condition not widely known, and frequently misdiagnosed. Spranger et al.(1) in their five cases reported that one patient was given various diagnoses including rheumatoid arthritis, polymyositis, endochondral dysosto-sis and Morquio disease, while another received the diagnosis of endochondral dysostosis. Our patient also reported to us with diagnoses of rickets, rheumatoid arthritis and MPS having been made elsewhere. Normal calcium, phosphorus and alkaline phospaha-tase, and absence of characteristic changes of rickets on X-rays of wrists and ankles excluded rickets, while a normal ESR, negative rheu-matoid factor, absence of fever, pain and tenderness ruled out rheumatoid arthritis. In view of the coarse facial features, short stature and skeletal abnormalities and normal IQ, Morquio disease was a likely possibility and a skeletal survey was ordered to look for bony changes of MPS. The X-rays however showed up the pathognomonic changes of PPAC(9,10). In view of the difficulty in early clinical diagnosis of the disease, it is imperative that radiologists be familiar with radiographic features of the disease in order to avoid needless investigations and trials of medication. The characteristic radiographic features are narrow joint space with wide metaphyses and flat epiphyses. Femoral heads are enlarged and acetabulae are irregular. Platyspondyly with erosion of end plates is present. This condition mimics juvenile rheumatoid arthritis but bony changes show joint space narrowing, over-growth of epiphyses and osteopenia, which are not seen in PPAV. This condition should also be differentiated from spondyloepiphyseal dysplasia congenita, in which the extremities are normal; from spondyloepiphyseal dysplasia tarda, where the patient has a characteristic dorsal hump and spondylo-metaphyseal dysplasia in which epiphyses are not affected and joint stiffness is not present. Though our patient showed all the classic clinical and radiological features of PPAC, the facial dysmorphism was misleading. Such facies have not been reported in association with PPAC earlier. Another hitherto un-reported feature in our patient was the arm span being greater than the height. A reduction of measured standing height may occur due to flexion deformities at the hips; however the flexion at the hips was around 10 degrees and does not explain the marked difference of 5 cm between arm span and standing height. Contributors: RM carried out the clinical workup and also drafted the manuscript. She will act as the guarantor for the paper. SV was responsible for the radiological aspects and co-drafted the manuscript. Funding:
None.
|
![]()