|
|
Case Reports Indian Pediatrics 2001; 38: 89-92 |
||||||||
|
Juvenile Sandhoff Disease |
||||||||
|
Juvenile Sandhoff’s disease is a rare disease causing progressive neurologic dys-function beginning in late childhood. It is autosomal recessive in inheritance and is characterized by a deficiency of hexos-aminidase A and B enzymes(1). We report a case of Juvenile Sandhoff’s disease which is to our knowledge, the first report of the disease in an Indian patient.
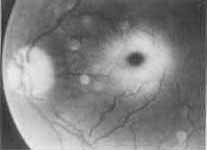
A fourteen-year-old girl presented to our department with a history of two episodes of generalized tonic clonic seizures in the last 3 months. She was born to a nonconsanguineous parents in a hospital. The antenatal and the postnatal periods had been uneventful. She had gone to school and had led an apparently normal life till about 9 years of age when she was noticed to fatigue easily and did not seem as energetic as other children. Over the past two years her father had noticed that she tended to stagger while walking. There was no history of similar illness in her family. On examination she was a young girl with a blood pressure of 120/70 mm/kg. She had a Marfanoid habitus. Her height was 145 cm. Her arm span was 150 cm. The wrist and thumb signs for a Marfanoid habitus were positive. She had a high arched palate. However other systemic and ocular features of Marfan syndrome were lacking. Nervous system examination revealed that her higher mental functions were normal. Her ophthalmic fundii revealed bilateral cherry red sport (Fig. 1) which was confirmed by an ohthal-mologist. She also had nystagmus on looking to the right and left. The sensory and motor system examinations were normal. Ankle jerks were sluggish, however the other reflexes were normal. She had dysdiadochoikinesia, rebound phenomenon and a positive finger nose test. Her gait was ataxic and the Rhomberg sign was negative. She did not have visceromegaly. Other systemic examination was normal.
Investigations revealed a normal hemo-gram, serum electrolytes and renal function. Her electroencephalogram revealed a normal pattern. Slit lamp examination did not reveal lens dislocation or any feature of Marfan syndrome or homocystinuria. Electromyo-graphy and nerve conduction studies were performed. These suggested a peripheral neuropathy involving both sensory and motor components. The bone marrow examination revealed no abnormal cells. Echocardiogram showed no cardiovascular features of Marfan syndrome. The differential diagnosis of bilateral cherry red spots with a progressive neurologic dysfunction included sialidosis and GM2 gangliosidosis. Sialidosis is characterized by myoclonic jerks and occurs due to neuraminidase deficiency. It was ruled out by the quantitative assesment of neuraminidase activity in leukocytes(2) using fetuin as the substrate with the enzyme prepared by the techniques of Varatharajan et al.(3) In addition sialic acid released from glyco-proteins and glycopeptides in a 24 hour urine sample was also estimated. A possibility of GM2 gangliosidosis was entertained and a quantitative estimation of Hexosaminidase A and B activity in leukocytes was performed using a technique described by Basu et al.(4). No Hexosaminidase A and B activity could be detected. Hence, a diagnosis of Juvenile Sandhoff’s disease was made.
Sandhoff disease is a storage disorder. The absence of hexosaminidase enzymes in this disease lead to the accumulation of GM2 gangliosides in neural tissue. Juvenile Sandhoff disease belongs to a wider spectrum of disorders loosely clumped under the term "GM2 gangliosidoses". The most well known amongst them is Tay Sachs disease where only hexosaminidase A is absent. In contrast both hexosaminidase A and B are absent in Sandhoff disease. The features of both Sandhoff and Tay Sachs disease are largely similar except for the differences in bio-chemical enzyme deficiency patterns(1). Hexosaminidase A and B are glycoprotein enzymes that have two subunits–alpha and beta. The beta subunit is common to both hexosaminidase A and B. This beta subunit is coded by a gene on chromosome 5. Mutations of this gene result in abnormality of both the enzymes manifesting as Sandhoff disease. These allelic mutations are diverse and can result in varying residual of the enzymes. This can explain certain variations in clinical presentation(1). Thus, Sandhoff disease can manifest in infancy or late childhood. In the latter, it is termed juvenile Sandhoff’s disease. The late and milder presentation of symptoms in our case is probably due to mutations which are different from those occuring in the classical infantile form. The earliest Indian case series of gangliosidosis was from Vellore(5). An etiologic study(5) of children with mental retardation showed that 14.5% of these cases were attributable to neurolipidosis. Juvenile Sandhoff disease is rare(1), but there have been several case reports of this illness(6,8). However, to our knowledge, this is the first report of Juvenile Sandhoff disease in an Indian subject. All GM2 gangliosidoses are lysosomal storage diseases in which the enzyme deficiency is present in all cells. So, why is there a prediliction for certain tissues? The most simple solution seems to be that gangliosides are produced only by nervous tissue. Hence, they will accumulate only in them, even though the enzyme is deficient in all cells. This would explain preferential ganglioside accumulation in neural tissue(1). The clinical manifestations can vary depending on the genetic defect. McLeod et al.(6) proposed the term "Juvenile" Sandhoff disease in a 10-year-old boy with ataxia, spasticity and psychomotor retardation with total hexosaminidase deficiency. The authors suggested that these patients with Juvenile Sandhoff disease have a benign protracted illness, while the infantile form of Sandhoff disease is characterized by seizures, retinal degeneration and rapid deterioration. The occurrence of the disease in adult siblings presenting predominantly as spinocerebellar ataxia has has been reported(6,7). Bilateral cherry red spots are not as common in Juvenile Sandhoff Disease when compared to Tay Sach’s Disease(1). To our knowledge, the occurrence of a Marfanoid habitus associated with Juvenile Sandhoff disease is unique to our patient and is hitherto unreported. The course of the disease is that of relentless and ultimately fatal progression of neurologic disease. There is no definitive treatment. A variety of innovative therapies including hexosaminidase infusions and leuko-cyte transfusions have been tried. Bone marrow transplantation has also been attempted. However, none of these have provided a cure(1). Gene therapy offers hope for the future. For the present, preventive genetic counselling and the supportive treat-ment of affected individuls are the only options. Recently a new drug called N-butyl-deoxynojirimycin has been found to delay onset and progression of Sandhoff disease in animal models(9). It inhibits the synthesis and storage of accumulating lipid and is a potential therapeutic strategy for other lysosomal storage diseases as well. If future trials establish the efficacy of this drug, it could offer hope to patients afflicted with this incurable illness.
The authors are grateful to Mrs. Rebecca Cherian, Biochemist, Department of Neuro-chemistry, CMC Hospital, Vellore for her valuable contribution in carrying out the biochemical assays. Contributors: UAG and SD were involved in the primary care of this patient. MS was the consultant in charge of this patient. All authors contributed to drafting of the manuscript. Funding:
None.
|
![]()