|
|
Case Reports Indian Pediatrics 2001; 38: 189-193 |
|||||||||||||||
|
Thoracic Epidural Neurilem-moma: A Rare Cause of Childhood Paraparesis |
|||||||||||||||
|
Spinal cord tumors commonly occur in the 4th to 6th decade of life with an incidence of 3 to 10 per 100,000 population and constitute a rare cause of childhood tumors(1,2). We report a case of a solitary intraspinal neurilemmoma, a tumor only occasionally reported in children(3).
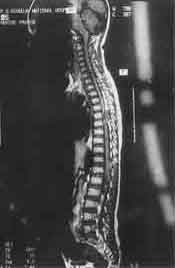
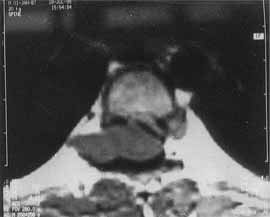
An eleven-year-old boy presented with a gradual onset lower limb weakness of one-month duration. The weakness began with slipping of the footwear and gradually progressed to the proximal muscles, making standing and walking difficult. It was associated with the development of stiffness in the lower limbs, hesitancy in micturition and constipation. There was no history of involve-ment of sensory faculty, higher functions or cranial nerves. Nor was there a history of fever, trauma, backache or tuberculous contact. On examination, the lower limbs had clasp knife rigidity and a power of grade 2/5. The deep tendon reflexes were exaggerated and ankle clonus was present bilaterally. The plantar response was extensor bilaterally. The superficial abdominal reflexes were absent and hypoaesthesia was noted below T-8 dermato-mal level. The examination of the spine revealed no abnormality. Rest of the clinical examination was non-contributory. There were no neurocutaneous markers. Further investigations were carried out with the clinical diagnosis of a compressive myelopathy. The plain roentgenogram of the spine was normal. The magnetic resonance imaging (MRI) scan of the spine revealed an isointense tumor on T1-weighted images (Fig. 1). The tumor was dumb-bell shaped, located at T6-T7 level and was seen to be extending into the extraspinal compartment through the neural foramina on the right side. It was noted to cause secondary effects in the form of compression and leftward displace-ment of the cord (Fig. 2).
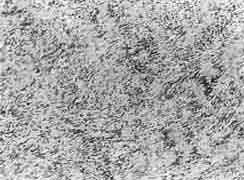
The patient underwent a D5-7 laminectomy and a complete excision of the tumor. At the surgery, the grayish red tumor was noticed to be arising from the posterior nerve root and was located epidurally without any intradural extension. It was a firm encapsulated mass, measuring 3.5 × 3 × 3 cm and was distorting and compressing the adjacent spinal cord. Histopathologically, it showed the character-istics of a neurilemmoma. The cellular tumor mass consisted predominantly of Antony A areas made up of closely packed spindle shaped cells with pale cytoplasm and ovoid central nuclei with pallisading of nuclei. Verocay bodies (longitudinally cut rows of nuclei separated by clear hyaline bands) were also seen (Fig. 3). Few areas of Antony B type with loosely arranged similar cells were seen too.
Following surgery, on day two of the postoperative period, patient’s power started improving. He made a remarkable recovery over next five days. At the end of which, his lower limb power improved to grade 4/5, sensations became normal and the sphincteric control was regained. He was advised physiotherapy and was discharged, but was subsequently lost for follow up.
Spinal cord tumors constitute an unusual cause of acquired childhood paraplegia. They account for only 20% of neuroaxial tumors in children(4). Hence, when a child presents to a physician with paraplegia, Guillain-Barre syndrome, poliomyelitis, spinal trauma, tuber-culosis of the spine and transverse myelitis are the commonly considered etiologies, depend-ing upon the clinical settings. In an infant congenital malformations like myelomeningo-coele and caudal regression syndrome constitute an all together different spectrum of etiology for paraparesis. Certain hereditary conditions like adrenoleukodystrophy and adrenomyelo-dystrophy may also present in older children with paraparesis. With the better availability of sensitive neuroimaging techniques, it is now possible to diagnose spinal cord tumors. In children astrocytoma, ependymoma, and neuro-blastoma are the commonest spinal tumors. Other reported spinal cord tumors in childhood include teratomas, dermoids and chordromas. These tumors are much more common than neurilemmomas. Neurilemmoma (also called schwannoma or neurinoma) is a benign tumor arising from the nerve sheath. It remains a tumor of an older age(3). It can arise as a primary intracranial tumor or as a spinal tumor(4). When presenting as a spinal cord tumor, neurilemmoma is usually intradural and extramedullary in loca-tion(5). However, when an intradural or extradural tumor extends into the paraspinal space, it gives rise to the classical dumb-bell shaped tumor, as in our case. Epidural neurilemmomas are uncommon and constitute only 15% of all the spinal neurilemmoma(6). Thoracic spine is the commonest site of occurrence as 50% of neurilemmomas occur in this region. Cervical region is the next commonly involved region. When located in the epidural space, the neurilemmoma is likely to present with radicular pain due to the irritation of the nerve root. Absence of such a pain in our patient is inexplicable and was probably responsible for his late presentation to us with manifestations of sensory and motor disturbances repre-senting the cord compression due to the enlargement of the tumor. These secondary events usually evolve over a period of months to years. However, in our patient, these events progressed rapidly spanning only a month. This could be indicative of a rapid growth of the tumor. Neuroimaging with MRI is the investiga-tion of choice in which the mass is seen as an isointense or slightly hypointense lesion in T1- weighted images. In T2-weighted and enhanced T1-weighted images, a target pattern with a peripheral hyperintense ream and a central low intesity area is seen(7). These changes correspond to the histological pattern of the peripheral myxomatous tissue and a central fibrocollagenous tissue. A neurofibroma is the closest differential diagnosis. However, MRI may not distinguish neurilemmoma from a neurofibroma and a neurilemmoma with cystic, hemorrhagic or necrotic degeneration may mimic malignant nerve sheath tumor on MRI. When MRI is not available, CT scan with myelography is a suitable alternative(2,8). On a histopathological examination, the neurilemmoma shows a spatial arrangement of the tumor cells into classical Antony A and B areas (vide supra). A characteristic feature, known as pallisading of nuclei, seldom encountered in the acoustic neurilemmoma, is often pronounced in the spinal neurilemmomas as evident in our case too. The tumor may also show small cysts due to the previous hemorr-hages into the tumor. Electron microscopy of neurilemmoma reveals presence of character-istic cytoplasmic processes coated with an electron dense basement membrane with Luse bodies in the matrix(3). Rare case of pigmented, glandular and pseudoglandular schwannoma have been described(3). Malig-nant change, though reported in literature, is rare(2). Surgical excision is the treatment for neurilemmomas and generally, complete excision is possible. The prognosis generally depends on the pre-surgical status and the extent of surgical excision possible. An in-complete excision is associated with a risk of the recurrence. In such cases, the risk of recurrence correlates with mitotic index(9). Brdu index, which depends on the number of cells in S-phase, is also used in prognostica-tion(10). Rapid onset progressive neurological deterioration and an inability to achieve complete excision of the tumor are associated with a poor prognosis with ambulation possible only in 35% of those who are bed ridden(11). However, despite rapid progression, presence of a significant motor deficit and a functional disability to the extent of being bed ridden, our patient showed remarkable recovery due to the complete excision of the mass. This emphasizes the need for an early diagnosis on the part of the treating physician and an early referral to a skilled neurosurgeon. Contributors: RCP did the collection of the data, drafting of the manuscript and will act as the guarantor. SBB helped in the revision of the manuscript. DRS assisted in the collection of the data and drafting of the manuscript. DP helped in the revision of the manuscript and provided the photomicrograph. JRK helped in revision of the manuscript.
Funding:
None.
|
![]()