|
|
Case Reports Indian Pediatrics 2000;37: 1373-1376 |
|||||||||
|
Engelmann’s Disease with Cardiomyopathy |
|||||||||
|
[email protected] Manuscript received: November 24, 1999; Initial review completed: January 18, 2000; Revision accepted: June 2, 2000. Engelmann’s disease is a rare condition with usual onset in childhood. The presenta-tion is mainly with muscular weakness and waddling gait. Radiologically, it is a prog-ressive diaphyseal dysplasia extending from the center of tubular bone towards the peri-phery. Diverse clinical presentations have been described in the literature. We report a case of Engelmann’s disease that had dilated cardiomyopathy. This association has never been previously described, to the best of our knowledge.
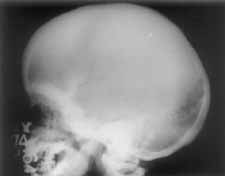
The girl was first brought to the hospital at the age of one year with complaints of severe pallor and inability to stand. She was a product of consanguineous marriage and was born at the hospital as a normal vaginal delivery with a birth weight of 2.7 kg. Ante-natal, natal and immediate postnatal history were non contributory. She had delayed developmental milestones. The physical growth was retarded as well. As hemogram and bone marrow did not provide any definite diagnostic clue and the child did not respond to hematinics, a skeletal survey was done which showed typical features of Engelmann-Camurati disease. Child was initially trans-fused with blood and later put on a two weeks course of prednisolone that led to improve-ment in hemoglobin. Subsequently, the child continued to suffer from recurrent episodes of anemia, easy bruisability, difficulty in walk-ing and respiratory infections needing several admissions and follow-up in the outpatient department. Child was treated with anti-biotics, bronchodilators, multiple blood trans-fusions and other supportive therapies includ-ing hematinics and multivitamins. As a repeat course of steroids led to severe respira- tory infection, the steroid were not tried subsequently. At the age of 4 years, she developed occasional episodes of edema over face and legs associated with tachypnea and decreased urinary output. Physical examination was suggestive of congestive cardiac failure. Chest X-ray revealed mild cardiomegaly EKG showed sinus tachycardia with no segment abnormalities. Echocardiography showed all chambers to be dilated with global hypo-kinesia of left ventricular wall and an ejection fraction of 40%. Features were suggestive of dilated cardiomyopathy. She was treated with decongestive measures, packed red blood cells and angiotensin II inhibitors regularly. Presently, the child is eight year old and active, though she walks slowly with a broad-based gait. She is thin and weighs 15 kg with a height of 102 cm and head circumference of 49.5 cm. She has thin lower limbs and reduced muscle mass. She has mildly tender thighs and calves. Echocardiography done recently has shown marked improvement in left ventricular function and an ejection fraction of 70%. The complete blood counts and blood chemistry are normal except for mild anemia (10.2 g/dl; normocytic to microcytic hypo-chromic peripheral smear) and a chronically elevated ESR. Skeletal survey of the child revealed markedly thickened skull base (Fig. 1) with sclerosis and mild thickening of the skull vault. The long and short tubular bones were thicknened in fusiform manner with diaphyseal, endosteal (Fig. 2) and perio-steal new bone formation. The metaphyses and epiphyses of tubular bones were normal. Generalized increase of bone density was additionally present. Clinical and radiological features are consistent with a diagnosis of Engelmann’s disease.
Engelmann-Camurati Disease (EC Disease) is a rare disorder affecting various systems. It has an autosomal dominant (AD) mode of transmission with variable pene-trance and expressivity(1). Approximately, 170 cases of this disorder had been reported till 1994(2). We have been able to collect 38 more cases from the literature since 1994 making the total to 208 cases. This disease has a usual onset in childhood with waddling gait, muscular weakness, extremity pain and asthenic body habitus. Muscle mass is often decreased in these children. A low muscle carnitine has been reported(3). A variety of eye disorders have been noticed in patients with this disease. The manifestations in the eye include diplopia, periorbital edema and ecchymosis(4). Also reported are optic atrophy, exophthalmos and tortuosity in the retinal veins(2). Multiple cranial nerve palsies can occur in association with the changes in the skull and this has been demonstrated on MRI(5). These patients may suffer recurrent episodes of bone marrow hypoplasia, chroni-cally increased ESR and recurrent infections. The infections are a manifestation of immunological abnormalities like impaired function of CD4+ cells. Radiology plays a vital part in the diag-nosis of this disorder. Progressive sclerosis of diaphyses of long bones in the absence of metaphyseal involvement is a characteristic feature. This leads to increased bone density in long and tubular bone. The excess deposi-tion of bone is both endosteal and periosteal. Histologically, there are features of rapid new bone formation, woven osteoid and lack of development of Haversian system in the bones(6). Skull is frequently involved in this disease. Scelortic changes in the skull with occasional involvement of vault and facial bones may be seen(2). Frontal and occipital bossing and frontal bone absorption lacunales have been described(7). Due to new bone formation in the skull base, the foramina of various cranial nerves and vessels may be progressively obliterated. The involved skele-ton reveals increased bone activity on nuclear bone scans(8). Our patient had almost all the classical manifestations of this disorder including pre-sentation in the form of myopathy, waddling gait, malnutrition, and recurrent episodes of infections. She had a history of severe anemia requiring multiple blood transfusions. Review of past medical records was compatible with a diagnosis of anemia due to bone marrow hypoplasia. Exophthalmos, reduced muscle mass, pallor and hepatosplenomegaly were present on clinical exam. The skeletal manifestations were characteristic. Therefore, the diagnosis of E-C disease was not difficult in the present case. She additionally had dilated cardiomyopathy, proven on echo-cardiography. This is the first report of a child with Engelmann’s disease with coexistent dilated cardiomyopathy. Differential diagnosis of Engelmann’s disease on the skeletal survey includes dia-physeal dysplasia and severe anemia(9), osteopetrosis, infantile cortical sclerosis and Ribbing’s disease. Absence of metaphyseal involvement and clinical feature differentiate it easily from osteopetrosis. Infantile cortical selerosis occurs in much younger age group and tends to involve clavicles, mandible and ribs. A radiographically similar disorder "Ribbing’s disease" may be very difficult to differentiate from this condition and accord-ing to some authors, it may be a milder form of Engelmann’s(2). Cardiac involve-ment has been reported in Ribbing’s disease(10). How-ever, presence of neuro-muscular dystrophy and malnutrition, as seen in our patient, are features of Engelmann’s disease and differ-entiate it from the Ribbing’s disease. Contributors: PC followed up the case, drafted the paper and will act as guarantor of the paper. VB and BB participated in drafting the paper. DG carried out and interpreted the radiological investigations. Funding: None.
|
![]()