|
|
|
Indian Pediatr Suppl 2009;46: S48-S54 |
 |
Markers of Fetal Onset Adult Diseases |
|
Latha Nair, MKC Nair and DS Chacko
From Child Development Centre, Medical College,
Thiruvananthapuram 695 011, Kerala, India.
Correspondence to: Dr MKC Nair, Professor of Pediatrics
and Clinical Epidemiology and,
Director, Child Development Centre, Medical College, Thiruvananthapuram
695 011, Kerala. India.
E-mail: [email protected]
|
Abstract
The "fetal origins hypothesis", proposes that
non-communicable diseases including coronary heart disease, type 2
diabetes and hypertension originate through the responses of a fetus to
undernutrition, that permanently change the structure and function of
the body. Associations between low birthweight and disease in later life
have been widely studied in Europe and the USA. Studies in southern
India have shown that babies who are short and fat tend to become
insulin deficient and have high rates of non-insulin dependent diabetes.
These findings have important public health implications as it suggests
that associations with body size at birth underestimate the contribution
of intrauterine develop-ment to later disease, and also, that while the
primary prevention of coronary heart disease and non-insulin dependent
diabetes may ultimately depend on changing the body composition and
diets of young women. Therefore, more immediate benefit may come from
preventing imbalances between prenatal and postnatal growth among
children. The basic premise of the thrifty gene hypothesis is that
certain populations may have genes that determine increased fat storage,
which in times of famine represent a survival advantage, but in a modern
environment result in obesity and type 2 diabetes. The fetal origins
theory is of greatest relevance to the developing world and the
implications of this work for global health are enormous. To reduce
chronic diseases, we need to understand how the human fetus is nourished
and how malnutrition changes its physiology and metabolism, so that
interventions be implemented to limit the damage. The challenge for the
next decade must be to discover the cellular and molecular mechanisms
giving rise to these associations. If this aim is accomplished, it might
be possible to devise strategies to reduce the impact of these disabling
chronic and expensive diseases.
Keywords: Barker hypothesis, Fetal origins hypothesis,
Non-communicable diseases, Thrifty gene hypothesis.
|
|
The core of the theory of fetal origins of
disease is that nutritional deprivation of the fetus during critical
periods of development forces the baby to resort to adaptive survival
strategies, which entail a resetting of the normal course of metabolic,
physiological, and anatomical development(1). These adaptations become
maladaptive if the organism encounters contrasting nutritional
circumstances in later life. It has also become clear that maternal
constraint must have a central role in fetal programming. Under such
circumstances, maternal uterine constraint becomes a dominant regulator of
fetal growth. The proponent of "fetal origins hypothesis" is a British
epidemiologist David Barker. The fetal origin, hypothesis was developed by
linking records of births in the early 20th century with health in later
life from the Hertfordshire records(1-10).
Associations between low birthweight and later disease
have been widely replicated in studies in Europe and the
USA(11-13). The association between low birth weight and coronary artery
disease has been confirmed in studies of men in Sweden(11), Helsinki,
Finland, and South Wales(14), and among 80 000 women in the American
nurses study(12). The fetal origins theory is of greatest relevance to the
developing world, and the implications of this work for global health are
enormous(15). Studies in southern India have shown that babies who are
short and fat tend to become insulin deficient and have high rates of
non-insulin dependent diabetes(16). These findings were similar to those
seen in Pima Indians and also with observations in Sheffield that showed
an association between abdominal circumference at birth and death from
coronary heart disease(17). Shortness and fatness are thought to be the
result of maternal hyperglycaemia, with consequent imbalance in the supply
of glucose and other nutrients to the fetus. Studies in Preston showed
that babies whose placentas are disproportionately large in relation to
their own weight tend to have raised blood pressure (18).
These findings have important
public health implications, as it suggests that associations with body
size at birth underestimate the contribution of intrauterine development
to later disease. While the primary prevention of coronary heart disease
and non-insulin dependent diabetes may ultimately depend on changing the
body composition and diets of young women, more immediate benefit may come
from preventing imbalances between prenatal and postnatal growth among
children. Many chronic disorders that manifest later in life may be
related to two seemingly opposing factors potentially present early in
life; (i) poverty (i.e, malnourished mothers give birth to
malnourished infants with low birth weight [LBW]), and (ii)
prosperity (exposure of an infant with LBW phenotype to a high caloric
diet). These factors contribute to the biological phenomenon of
developmental plasticity, or the ability of a genotype to produce multiple
forms and behaviors in response to environmental conditioning(19). The
Fetal origin hypothesis is summarized in Fig.1(19).
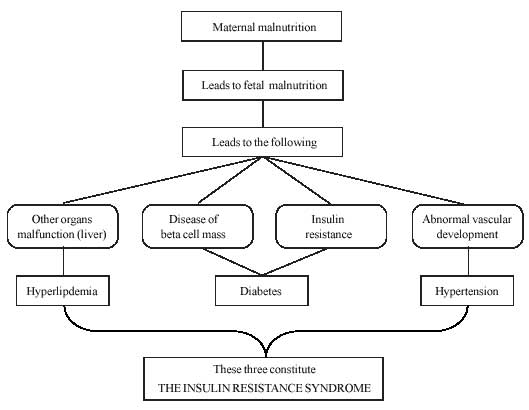 |
|
Fig. 1
Fetal origins hypothesis |
Four birth phenotypes associated
with later disease have been identified; (a) babies who are thin at
birth; (b) babies who are short at birth; (c) babies short
and fat at birth, and (d) babies born with a large placenta(19).
Babies that are thin tend to be insulin resistant as children and adults,
and are therefore liable to develop the insulin resistance syndrome(20).
It could be that the thin baby has adapted to under-nutrition through
endocrine and metabolic changes. Babies that are short in relation to
their head circumference, and babies that have a reduced abdominal
circumference, tend to have persisting abnormalities of liver function,
including raised serum LDL cholesterol and plasma fibrinogen
concentrations(21,22). Babies that have a small abdominal circumference in
relation to their head circumference can result from "brain sparing"
circulatory adaptations by which cardiac output is diverted to the brain
at the expense of the trunk(23).
The Thrifty Genotype and the Thrifty Phenotype
The prospective studies by Yajnik,
et al.(24) in Pune in India are therefore notable and deserve
attention. Caroline Fall from David Barker’s Medical Research Council
group in Southampton along with Yajnik and his team have used
anthropometric measurements of babies to describe their morphology at
birth(25). According to Yajnik, maternal uterine constraint becomes a
dominant regulator of fetal growth in order to protect the mother from
having to deliver an inappropriately large baby. In rural villages mothers
average about 44 kg in mid-gestation, with a height of 152 cm and body
mass index of 18 kg/m2(25). This leads to fetal malnutrition, which may be
a major component in the susceptibility to coronary heart disease and
non-insulin-dependent diabetes.
In the words of J V Neel, the initial proponent of the
thrifty genotype hypothesis, the
thrifty genotype is "rendered detrimental by progress" and leads to high
rates of metabolic syndrome and type 2 diabetes(26). This has provided an
opportunity to assess the state of the hypothesis and consider its
implications for future research and policy(27,28). Along with inadequate
fetal nutrient supply, other explanations, including the operation of
genetic factors and programming of certain endocrine axes, have also been
put forward to explain the origin of these non-communicable diseases and
the epidemiological associations(14). In relation to insulin action and
diabetes, Hales and Barker have described this phenomenon as the "thrifty
phenotype"(29-31). The basic premise of the thrifty gene hypothesis is
that certain populations may have genes that determine increased fat
storage, which in times of famine represent a survival advantage, but in a
modern environment, result in obesity and type-2 diabetes(32,33).
Intrauterine growth retardation (IUGR) or clinically abnormal thinness at
birth strongly predicts the subsequent occurrence of hypertension,
hyperlipidemia, insulin resistance, type 2 diabetes and ischemic heart
disease.
The concept of fetal programming during development has
been proposed to explain these findings. Fetal undernutrition, during
middle gestation in particular, raises the risk of later disease by the
programming of blood pressure, cholesterol metabolism, blood coagulation
and hormonal settings(1). One third of Indian babies have a low
birthweight, on average they weigh around 2.7kg. This makes them highly
susceptible to conditions mentioned earlier when they are older. The
thinness of Indian babies is due to poor muscle mass and small abdominal
viscera and is due to the "thrifty phenotype" of Indian babies, which
enhanced survival in subsistence conditions in the past, but becomes
detrimental in a modern context of plentiful food and reduced physical
activity(34). Yajnik also showed that in the babies of urban mothers in
Pune, insulin concentrations in the
blood of the cord seem raised compared with British babies and were
correlated with subscapular skin-fold thickness(35). Indian babies are
much smaller than those in Southampton in all respects except measures of
body fat-especially central fat as judged by the subscapular skin-fold
thickness. They describe this as the "thin-fat" baby syndrome and believe
that it shows that the excess visceral adiposity of most asian adults can
be traced back to the neonate(24,36). In a study conducted in Mysore,
India, it was found that low birthweight men and women were insulin
resistant and that coronary heart disease and its risk factors were linked
to features of insulin resistance syndrome(16,37).
The obvious
response to the "small baby predicts later disease" paradigm is to propose
dietary supplementation of mothers to produce larger babies. We should act
to prevent retarded fetal growth in mothers whose diet is so poor as to
limit the baby’s expected growth trajectory in relation to its parental
and genetic inheritance, and to the maternal uterine environment(38). But
it has been seen that low birth weight followed by catch-up growth was an
important risk factor for later disease, over and above low birth weight
itself(29). Therefore, if we already have short thin-fat mothers producing
small thin-fat babies, should we really be feeding them more? This is a
tricky question. The answer is probably "no", because this results in
augmented fetal growth which will be out of harmony with the baby’s
inheritance and future growth patterns. The resolution of this conundrum
will require focused investment in international studies on the regulation
of early human growth and development (38).
Intrauterine Programing
In conditions of undernutrition,
a genotype conferring insulin resistance would be preferentially selected
during evolution because this genotype would increase survival among small
babies, who would otherwise have a high perinatal mortality. This
phenomenon has been referred to as "the surviving small baby
hypothesis"(14,17). On the basis of this finding, it has been suggested
that this gene, which increased birth weight, might enhance perinatal
survival and perhaps paradoxically increase susceptibility to type 2
diabetes(14).
Several genes have already been identified as
candidates for the thrifty genotype, including those encoding proteins of
the insulin-signaling and leptin pathways, as well as intermediary fat
metabolism. Particular interest lies in the peroxisome-proliferator
activated receptors. According to Joffe, et al.(33), an innovative
approach might be to focus on the "mirror image" of the thrifty genotype -
congenital lipoatrophic diabetes mellitus, whose molecular defect remains
enigmatic. They conclude that the genetic basis of the thrifty genotype
probably derives from the multiplicative effects of polymorphisms at
several sites, rather than a single regulatory abnormality(33). More
recently the molecular biology of this
process is emerging as a fascinating conflict between maternal and
paternal influences that involves a range of imprinted genes, especially
insulin-like growth factor-2 and its receptors(39). The protein
‘32-33 split pro-insulin’ is now identified as a marker of impaired
pancreatic beta cell function. This is a biologically inactive precursor
of insulin. This is found to be elevated in IUGR and may have a role in
future development of type-2 diabetes. It has therefore become apparent
that it is the disharmony between fetal growth and later growth rates that
seems to be the best predictor of the later pathology(2,40). There
is therefore a clear need to study interactions between genes and nutrient
supply in utero.
Hormones have also been implicated to
regulate fetal growth and development of individual fetal tissues, and
they have a central role in intrauterine programming. Nutritional
challenges that reduce fetal nutrient availability lower anabolic hormones
[e.g. insulin, insulin-like growth factor (IGF)-I, thyroxine (T4)]
and increase catabolic hormone concentrations [e.g. cortisol,
catecholamines, growth hormone (GH)]. Challenges that increase the fetal
nutrient supply raise anabolic and reduce catabolic hormone levels in the
fetal circulation. Certain patterns of intrauterine growth, particularly
growth retardation, can be related to specific postnatal outcomes.
Hormones have a central role in intrauterine programming, and insulin,
insulin-like growth factors, thyroxine and the glucocorticoids act as
nutritional and maturational signals and adapt fetal development to
prevailing intrauterine conditions, thereby maximizing the chances of
survival both in utero and at birth. However, these adaptations may have
long-term sequelae(41).Of the hormones known to control fetal development,
it is the glucocorticoids that are most likely to cause tissue programming
in utero. They are growth inhibitory and affect the development of all the
tissues and organ systems most at risk of postnatal pathophysiology when
fetal growth is impaired. Their concentrations in utero are also elevated
by all the nutritional and other challenges known to have programming
effects. Glucocorticoids act at cellular and molecular levels to
alter cell function by changing the expression of receptors, enzymes, ion
channels and transporters. They also alter various growth factors,
cytoarchi-tectural proteins, binding proteins and components of the
intracellular signaling pathways. Glucocorticoids act directly on genes
and indirectly through changes in the bio-availability of other hormones.
These glucocorticoid-induced endocrine changes may be transient or persist
into postnatal life with consequences for tissue growth and development
both before and after birth. In the long term, prenatal glucocorticoid
exposure can permanently reset endocrine systems, such as the
somatotrophic and hypothalamic–pituitary–adrenal axes, which, in turn, may
contribute to the pathogenesis of adult disease. Endocrine changes may,
therefore, be both the cause and the consequence of intrauterine
programing(41). Glucocorticoids act at cellular and molecular
levels to alter cell function by changing the expression of receptors,
enzymes, ion channels and transporters. They also alter various growth
factors, cytoarchi-tectural proteins, binding proteins and components of
the intracellular signaling pathways(41).
Influences that
Act in Postnatal life
Influences that
act in postnatal life add to the effects of low birth weight. The highest
prevalence of non-insulin dependent diabetes is found in people who had
low birth weight but were obese as adults. The highest death rates from
coronary heart disease occurred in men who were thin at birth but had
accelerated weight gain in childhood. We do not yet know whether this
association is because of the pathological effects of a high fat mass
persisting into adult life, deleterious effects of catch up growth, or the
intrauterine resetting of endocrine axes that control growth(10). It is
not known why catch-up growth is detrimental, but one speculation is that
fetal growth restriction leads to reduced cell numbers, and subsequent
catch-up growth is achieved by overgrowth of a limited cell mass. A
possible link between catch-up growth and coronary heart disease is that
it reflects persisting changes in secretion of hormones, including
insulin, insulin-like growth factor 1, and growth hormone, which are
established in utero in response to undernutrition and influence
both childhood growth and coronary heart disease. It is also possible that
if they develop a high body mass in childhood they have a
disproportionately high fat mass(42).
Babies born in countries undergoing rapid transition
would face malnutrition in their intrauterine life and a state of relative
over nutrition in later life, which provides oppurtunities for ‘catch up’.
Babies which catch–up in body weight, fat and height are more insulin
resistant as children(43). It is always better to take steps to prevent
low birth weight babies being born rather than giving post natal
nutritional supplementation because it is more rewarding to avoid obesity
in those who were small at birth(44). An understanding of the mechanisms
regulating fetal development is
important and an improved understanding of these mechanisms will emphasize
new approaches to prevent diseases such as atherosclerotic vascular
disease and type 2 diabetes(14). If fetal development can be better
optimized, there is definitely the potential to reduce the escalating
impact of type 2 diabetes and atherosclerotic vascular disease.
Funding: None.
Competing interests: The findings and conclusions
in this Review article are those of the authors and do not necessarily
represent the views of the funding agency.
References
1. Barker DJP. Fetal origins of coronary heart disease.
BMJ 1995; 311: 171-174.
2. Barker DJP, Winter PD, Osmond C, Margetts B. Weight
in infancy and death from ischaemic heart disease. Lancet 1989; ii:
577-580.
3. Law CM, Shiell AW. Is blood pressure inversely
related to birth weight? The strength of evidence from a systematic review
of the literature. J Hypertension 1996: 14: 935-941.
4. Mckeigue PM, Shah B, Marmot MG. Relation of central
obesity and insulin resistance with high diabetes prevalence and
cardiovascular risk in South Asia. Lancet 1991; 337: 382-386.
5. Yajnik CS, Fall CHD, Vaidya U, Pandit AN, Bavdekar
A, Bhat DS, et al. Fetal growth and glucose and insulin metabolism
in four year old Indian children. Diabetic Med 1995; 12: 330-336.
6. Barker DJP, Osmond C, Simmonds SJ, Wield GA. The
relation of small head circumferance and thinness at birth to death from
cardiovascular diseases in adult life. BMJ 1993: 306: 422-426.
7. Stien CE, Fall CHD, Kumaran K, Osmond C, Cox V,
Barker DJP. Fetal growth and coronary heart disease in South India. Lancet
1996; 348; 1269-1273.
8. Gupta R. Gupta VP. Meta-analysis of coronary heart
disease in India. Indian Heart J 1996; 48: 241-245.
9. Barker DJP, Osmond C, Goding J, Kuh D, Wadsworth
MEJ. Growth in utero, blood pressure in childhood and adult life, and
mortality from cardiovascular disease. BMJ 1989; 298: 564–567.
10. Hales CN, Barker DJP, Clark PMS, Cox LJ, Fall CHD,
Osmond C, et al. Fetal and infant growth and impaired glucose
tolerance at age 64. BMJ 1991; 303: 1019-1022.
11. Leon DA, Koupilova I, Lithell HO, Berglund L,
Mohsen R, Vagero D, et al. Failure to realise growth potential in
utero and adult obesity in relation to blood pressure in 50 year old
Swedish men. BMJ 1996; 312: 401-406.
12. Rich-Edwards JW, Stampfer MJ, Manson JE, Rosner B,
HanKinon SE, Colditz GA, et al. Birth weight and risk of
cardiovascular disease in a cohort of women followed up since 1976. BMJ
1997; 315: 396-400.
13. Leon D, Lithell HO, Vagero D, Koupilova I, Mohsen
R, Berglund L, et al. Reduced fetal growth rate and increased risk
of death from ischaemic heart disease: cohort study of 15000 Swedish men
and women born 1915-29. BMJ 1998; 317: 241-245.
14. Byrne CD, Philips DI. Fetal origins of adult
disease: epidemiology and mechanisms. J Clin Pathol 2000; 53: 822-828.
15. World Health Organization/UN Food and Agriculture
Organization. Diet, nutrition and the prevention of chronic diseases.
Geneva: WHO; 2003.
16. Fall CH, Stein CE, Kumaran K, Cox V, Osmond C,
Barker DJ, et al. Size at birth, maternal weight, and type 2
diabetes in South India. Diabet Med 1998; 15: 220-227.
17. McCance DR, Pettitt DJ, Hanson RL, Jacobsson LTH,
Knowler WC, Bennett PH. Birth weight and non-insulin dependent diabetes:
thrifty genotype, thrifty phenotype, or surviving small baby genotype? BMJ
1994; 308: 942-945.
18. Barker DJP, Bull AR, Osmond C, Simmonds SJ. Fetal
and placental size and risk of hypertension in adult life. BMJ 1990; 301:
259-262.
19. Barker DJP. Mothers, babies and health in later
life. Edinburgh: Churchill Livingstone; 1998.
20. Phillips DIW, Barker DJP, Hales CN, Hirst S, Osmond
C. Thinness at birth and insulin resistance in adult life. Diabetologia
1994; 37: 150-154.
21. Barker DJP, Martyn CN, Osmond C, Hales CN, Fall
CHD. Growth in utero and serum cholesterol concentrations in adult life.
BMJ 1993; 307: 1524-1527.
22. Martyn CN, Meade TW, Stirling Y, Barker DJP. Plasma
concentrations of fibrinogen and factor VII in adult life and their
relation to intra-uterine growth. Br J Haematol 1995; 89: 142-146.
23. Dicke JM. Poor obstetrical outcome. In: Pauerstein
CJ, ed. Clinical Obstetrics. New York: John Wiley and Sons; 1987.
p.421-439.
24. Yajnik CS. Interactions of perturbations in
intrauterine growth and growth during childhood on the risk of adult-onset
disease. Proc Nutr Soc 2000; 59: 257-265.
25. Yajnik CS, Fall CH, Coyaji KJ, Hirve SS, Rao S,
Barker DJ, et al. Neonatal anthropometry: the thin-fat Indian baby.
The Pune Maternal Nutrition Study. Int J Obesity 2002; 27:173-180.
26. Neel JV. Diabetes mellitus: a "thrifty" genotype
rendered detrimental by "progress." Am J Human Genet 1962; 14: 353-361.
27. Barker DJB. Fetal and Infant Origins of Adult
Disease. London: BMJ Publishing Group; 1992.
28. Hales CN, Barker DJP. Type 2
(non-insulin-dependent) diabetes mellitus: the thrifty phenotype
hypothesis. Diabetologia 1992; 35: 595-601.
29. Osmond C, Barker DJ. Fetal, infant, and childhood
growth are predictors of coronary heart disease, diabetes, and
hypertension in adult men and women. Environ Health Perspect 2000; 108
Suppl 3: 545-553.
30. Barker DJ. The long-term outcome of retarded fetal
growth. Schweiz Med Wochenschr 1999; 129: 189-196.
31. Strauss RS. Effects of intrauterine environment on
childhood growth. Brit Med Bull 1997; 53: 81-95.
32. Chukwuma C Sr, Tuomilehto J. The ‘thrifty’
hypotheses: clinical and epidemiological significance for
non-insulin-dependent diabetes mellitus and cardiovascular disease risk
factors. J Cardiovasc Risk 1998; 5: 11-23.
33. Joffe B, Zimmet P. The thrifty genotype in type 2
diabetes: an unfinished symphony moving to its finale? Endocrine 1998; 9:
139-144.
34. Law CM, Barker DJP, Osmond C, Fall CHD, Simmonds
SJ. Early and abdominal fatness in adult life. J Epidemiol Comm Health
1992; 46: 184-186.
35. Yajnik CS, Lubree HG, Rege SS, Naik SS, Deshpande
JA, Deshpande SS, et al. Adiposity and hyperinsulinaemia in Indians
are present at birth. J Clin Endocrinol Metab 2002; 87: 5575-5580.
36. Frühbeck G, Gomez-Ambrosi J, Muruzabal FJ, Burrell
MA. The adipocyte: a model for integration of endocrine and metabolic
signaling in energy metabolism regulation. Am J Physiol Endocrinol Metab
2001; 280: E827-847.
37. Kumaran K, Fall CHD. Fetal origins of coronary
heart disease and hypertension and its relevance to India; Review of
evidence from the Mysore studies: Int J Diabetes 2001; 21: 34-41.
38. Adair LS, Prentice AM. A critical evaluation of the
fetal origins hypothesis and its implications for developing countries. J
Nutr 2004; 134: 191-193.
39. Kelsey G, Constancia M, Dean WL, Feil RP, Reik W.
Genomic Imprinting of Fetal Growth. In Fetal Programming: Influences on
Development And Disease In Later Life. O’Brien PMS, Wheeler T, Barker DJP,
eds. London: Royal Society of Obstetrics and Gynaecology Press; 1999. p.
73-84.
40. Forsén T, Tuomilehto J, Reunanen A, Osmond C,
Barker D. The fetal and childhood growth of persons who develop type II
diabetes. Ann Int Med 2000; 33: 176-182.
41. Fowden AL, Forhead AJ. Society for Reproduction and
Fertility: Endocrine mechanisms of intrauterine programming.
Reproduction 2004; 127: 515-526.
42. Eriksson JG, Forsen T, Tuomilehto J, Winter PD,
Osmond C, Barker DJP. Catch-up growth in childhood and death from coronary
heart disease: a longitudinal study. BMJ 1999; 318: 7-1; 427-431.
43. Yajnik CS. Fetal origins of adult disease: Where do
we stand? Int J Diabetes 2001; 21: 42-50.
44. Haffner SM, Miettinen H. Insulin resistance implications for type
II diabetes mellitus and coronary heart disease. Am J Med 1997; 103:
152-162. |
|
|
 |
|
