|
|
Case Reports Indian Pediatrics 2001; 38: 1041-1045 |
||
|
Sirenomelia Sequence
Associated with Craniorachischisis Totalis, |
||
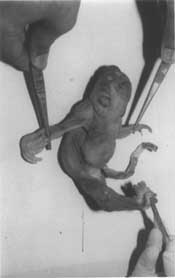
Sirenomelia is thought to occur in about 1 in 65,000 livebirth (Oxford Medical Database; Dysmorphology). In sirenomelia, lower limbs are fused together. Common associated malformations include absent external genitalia, imperforate anus, lumbo-sacral vertebral pelvic anbormalities and renal agenesis. Sirenomelia with craniorachischisis totalis (CT) is a rare fetal malformation. Only six cases have been reported in English liteature(1-5) and none yet from Asia. The case being reported here is unique for its association with undescribed cardiac abnor-malities (resembling a primitive heart) and a severe limb reduction defect. Some of the clinical findings of this case can not be explained by the axial mesodermal dysplasia theory alone, suggesting a possible coexist-ence of a vascular insufficiency. Case Report A 22 weeks fetus with multiple congenital malformations was sent to the Department of Medical Genetics, SGPGI, Lucknow for fetal autopsy so that diagnosis and counselling could be given to the couple. The fetus was born to a 22 years old primigravida mother and 30 years old father. It was a Muslim couple without history of consanguinity. The mother had an episode of upper respiratory tract infection (fever 103ºF) for 3-4 days without skin rash at 12th week of pregnancy. She had also received some unidentified homeopathic medicine in early pregnancy for nausea and vomiting. At 22 weeks of gestation, an ultrasound examination was carried out for decreased fetal movements and small for gestation uterine height. It revealed extreme oligo-hydramnios along with absence of cranial vault, spina bifida, absence of urinary bladder and sluggish fetal heart activity. The treating consultant terminated the pregnancy and a stillborn fetus with multiple congenital anomalies was referred to SGPGI (Fig. 1). The fetus weighed 167 g and was of unidentifiable sex. External examination showed craniorachischisis totalis (anence-phaly and total spina bifida aperta; Fig. 2) absence of pubis, definite buttocks, external genitalia, anus and urethra. There was a single fused lower limb ending in a bifid foot with six toes (three in each; Fig.1). The left upper limb was markedly shrunken with gradual tapering and ended at mid forearm (Figs. 1 and 2). Ears were low set and the palate was high arched. Necrogram showed an absence of vault bones. There were vertebral abnormalities, with ill defined fifth lumbar vertebra, sacrum and coccyx (severely hypoplastic and all fused together) in addition to scoliosis. Pelvic bones were ill formed and without pubis. There were two femorae and two tibiae (no fibulae). Femorae were fused at both ends. Left upper limb showed ill defined hypo-plastic small and incomplete humerus which ended at mid-shaft. Internal examination demonstrated absence of adrenals, kidneys, ureters, bladder, urethra, rectum, anal canal and internal genitalia including recognizable gonads. Heart showed a single atrium and ventricle, like a primitive heart. Histopathological examination revealed presence of testicular tissue (immature seminiferous tubules from approximately 1 mm length flat oval struc-tures obtained from the floor of a hypoplastic pelvis) and a single artery in the umbilical cord. Interphase fluorescent in-situ hybridiza-tion was carried out from formalin fixed liver and bone tissue(6). Decalcification steps pre-ceded release of cells from the bone. Chromosome X (centromers; green) and Y (centromere; red) specific directly labelled probes were used for this purpose (Vysis Inc., USA). It revealed presence of one copy of chromosome X and Y, respectively.
Discussion This case is very similar to one reported by Rodriguez and Palacios(5), with additional findings of a severe heart defect (primitive heart) and a severe left upper limb reduction. The only complication during pregnancy in the case was an episode of acute upper respiratory tract infection without skin rash at 12th week of pregnancy which is probably unrelated, as some defects like heart anomaly, sirenomelia and craniospinal defect, originate much earlier in pregnancy. Sirenomelia, characterized by single fused lower limb, includes multiple defects of contiguous organs such as uro-genital system, rectum and lower vertebrae, which constitute a single developmental field. This sequence is known to be associated with noncontiguous ano-malies, such as CNS defect and radial dysplasia, which do not lie in the same field. We came across two more cases of sireno-melia without any CNS or radial defects over a period of last 3 years. The pathogenesis of Sirenomelia Sequence is uncertain. Some of the proposed theories include posterior axial mesodermal defect(7,8), vascular steal(9,10), teratogenic effect(3,11), axial mesodermal dysplasia sequence(12) and midline developmentat field defect(2). Some of the, manifestations are secondary to oligohydramnios (e.g., Potter sequence including pulmonary hypoplasia) and vascular insufficiency (e.g., radial hypo-plasia, limb reduction etc.) which are evident in our case. Absence of familial cases and discordance in monozygotic twin(13) make a genetic mechanism unlikely. The limb reduction (hypoplasia and gradual of left upper limb) in the case reported here indicates chronic vascular insufficiency as the cause since the soft tissue ended at mid forearm, whereas the bone at mid humerus. This is also supported by the findings of a primitive heart and two vessels umbilical cord. Similarly, Stevenson et al.(9), have suggested that if the vitelline vascular system in the umbilical cord does not regress (‘Vitelline Steal’) then these abnormalities can occur. The association of craniorachischisis totalis and sirenomelia indicates the existence of a defect right from early embryogenesis. Formation of the neural tube first occurs on day 22 at the level of somite 3; fusion proceeds both rostrally and caudally and the neural tube is closed by day 26 whereas lower limb buds are recognizable between 28-32 days of developments(12). From an embryo-logical point of view, a condition that combines cephalic (craniospinal rachischisis) and caudal defects (fused lower limbs along with pelvic, lower vertebrae and genitourinary organ anomalies), as in this case, are consi-dered to be example of axial mesodermal dysplasia sequence(7,8,12). A primary distur-bance in the axial mesodermal system may impair formation, elevation and approxima-tion of neural folds leading to craniospinal rachischisis(14). Similarly, sirenomelia may occur due to failure in lateralization of lower limbs secondary to mesenchymal deficiency of the caudal eminence(l2), i.e., axial mesoderm. The caudal eminence is an active seat of cell proliferation which contributes to the formation of notochord, vertebrae, lower limb buds, perineum, neural plate, neural cord, hind gut and blood vessels. Although an extensive disturbance of axial mesoderm is likely to occur in the formation of neural tube defect, a milder alteration of neuro-ectoderm can not be excluded in most sirenomelia as defect occurs in coordinated process of cell proliferation, migration and cell to cell adhesion. Contributors: AH diagnosed the condition, coordi-nated work-up, carried up interphase FISH and draft the paper; he will act as the guarantor of the paper. JP and VC carried out fetal autopsy and necessary investigations for diagnosis. SSA helped in drafting the paper alongwith some scientific input.
Funding:
None.
| ||
| References | ||
|
![]()