|
|
Case Reports Indian Pediatrics 2000;37: 1017-1020 |
||||
|
Niemann-Pick Disease: Ultrastructural Features |
||||
|
Niemann-Pick disease refers to a group of disorders that are clinically, biochemically and genetically heterogenous. The predominant phospholipid accumulated in this disorder is sphingomyelin(1). It is inherited as an autosomal recessive disorder. Niemann-Pick disease has been divided into two forms: Type-I is character-ized by severe sphingomyelinase deficiency and Type-II in which sphingomyelinase activity is normal or only moderately diminished(2). Each of this group has been further divided into three clinical forms: (a) Acute (presenting in early infancy), (b) Subacute and (c) Chronic (detected in adulthood). Originally thought to occur only in infants, it has since been found that manifestations of the disease may first appear as late as the second year of life and even later(3,4). Since 1957, atleast 25 cases of Niemann-Pick disease have been reported from India(5-7). The diagnostic criteria included the demonstration of characteristic foam cells and sphingomyelinase assay. Ultrastructural analysis of Niemann-Pick cells has not been reported from India so far. In our Institute, 12 cases of Gaucher disease have been diagnosed over the last 10 years. In comparison only one case of Niemann-Pick disease has been reported(6). We present another case in whom ultrastructural analysis was undertaken.
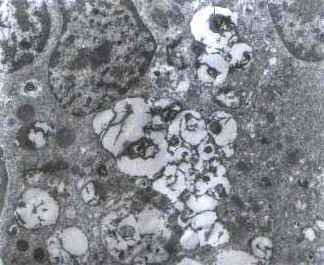
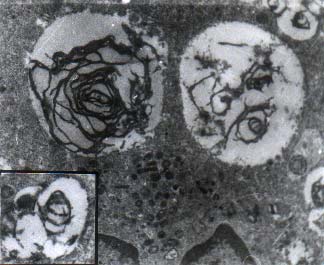
A 1½-year-old first born child of consangui-neous marriage was admitted in November, 1998, with the complaints of failure to thrive and delayed developmental milestones for one year. Physical examination showed normal pulse rate, blood pressure and respiratory rate. He had hepatomegaly of 8 cm below the right costal margin and splenomegaly of 6.5 cm below the left costal margin. At presentation hemoglobin (Hb) was 12.4 g/dl, total leukocyte count (TLC) was 16.4 ´ 109/L with a differential leucocyte count (DLC) of 41% polymorphs, 50% lymphocytes, 3% monocytes and 6% eosinophils. Platelet count was 240 ´ 109/L. RBC morphology was unremarkable. No nucleated red blood cells were seen. Bone marrow aspirate showed normo-cellular particles with a myeloid to erythroid ratio of 2.7:1 with normoblastic erythropoiesis and adequate representation of megakaryocytes. Differential count of the marrow showed blasts 1%, promyelocytes 3%, myelocytes 17%, meta-myelocytes 22%, polymorphs 30%, lymphocytes 18%, monocytes 3% and eosinophils 6%. Many storage cells were seen in bone marrow aspirate as well as trephine biopsy. These cells were large in size (25-50 mm), with eccentrically placed one to two nuclei and vacuolated cytoplasm. Cytochemical studies showed faint positivity for periodic acid schiff (PAS) stain. Electron microscopic study showed numerous storage cells with engorged secondary lysosomes (Fig. 1) which in turn contained many completely or incompletely formed myelin figures and occasional configurations resembling zebra body (Fig. 2). Retinal cherry red spot was not detected.
Part of cells or whole cells are regularly replaced in all tissues, during the course of normal growth, development and senescence. Sequential enzymatic degradation is required for breakdown of complex cellular constituents, taking place largely in secondary lysosomes. Lipid storage disorders are caused by absence of one of the lysosomal enzymes required for lipid degradation(3). There are increased levels of sphingomyelin and cholesterol in bone marrow cells, liver, spleen and brain. A characteristic ballooning of neurons in both grey and white matter occurs in central nervous system(8). The classic, infantile or A form of Niemann-Pick disease begins in infancy with signs of mental retardation, hepatosplenomegaly and sometimes lymphadenopathy. In about one half of patients, cherry red spot is seen(9). Most cases of the disease (approximately 85%) are of the infantile type(3). The diagnosis of lipid storage disorders may be considered in view of the clinical presentation of patient. However, the distinction between Gaucher's and Niemann-Pick disease can be made on the basis of light microscopic and ultrastructural features of the storage cells, and specific enzyme assays. Niemann-Pick cells are typically large foam cells, containing small lipid droplets. Presence of lipid droplets can be confirmed by phase microscopy, under polarized light or ultraviolet light. Typical appearance of the Gaucher cells is described as ‘crumpled tissue paper’. Ultrastructurally Gaucher cells reveal spindle or rod shaped membrane bound inclusion bodies consisting of numerous small tubules(3), whereas Niemann-Pick cells contain numerous engorged secondary lysosomes, with myelin figures and zebra bodies(10). Niemann-Pick disease as seen in India, seems to be similar in many aspects, to the disease reported in the Western literature. Out of the 26 cases detected in India so far (including this case report), 21 presented in infancy, hepatosplenomegaly was present in 26 and cherry red spot was detected in 3 cases only(5,7). Our case revealed numerous Niemann-Pick cells in the bone marrow, with characteristic ultrastructural features. The propositus demonstrates that the characteristic morphology of cells helps to differentiate between Niemann-Pick and Gaucher cells, and ultrastructural analysis (if undertaken) reveals typical features.
Fig. 1. Electron microphotograph of marrow sample showing many engorged lysosomes with myelin figures (Uranyl acetate and lead citrate stain ´ 4,500)
Fig. 2. Electron microphotograph showing myelin figure (´ 10,000) and Zebra body-inset (´ 15,000) (Uranyl acetate and lead citrate stain).
The authors gratefully acknowledge the help rendered by Dr. Amita Trehan (Department of Pediatrics) and Mr. H.S. Komal (Electron Microscopy Laboratory, Department of Histo-pathology).
|
![]()