M
ovement disorders are
conditions
characterized by involuntary postures and/or movements. It represents a common
presentation in pediatrics and is often a source of clinical and
diagnostic dilemmas [1]. Classically, movement disorders are
classified into hyperkinetic and hypokinetic disorders.
Hyperkinetic disorders are charac-terized by abnormal
involuntary movements and include dystonia, chorea, athetosis,
stereotypies, myoclonus, tics and tremor. Hypokinetic disorders
have in common a paucity of movements and include rare
conditions like parkinsonism [2]. Dystonia and chorea are the
most common forms of movement disorders. In many conditions,
multiple types of movement disorders co-exist and it may be
difficult to identify the type of movements.
There are no estimates on the prevalence of
movement disorders in children or their proportion amongst
pediatric presentations. The exact pathophysiology of movement
disorders is not well understood; however, evidence suggests the
involvement of either basal ganglia or cere-bellar circuits in
most of the conditions, which includes parts of thalamus and
cortex [3]. We, herein, cover common movement disorders with
emphasis on treatable conditions.
DYSTONIA
Dystonia is characterized by sustained or
intermittent muscle contractions causing abnormal, often
repetitive movements, postures, or both. Dystonic movements are
typically patterned (repeatedly involve the same group of
muscles), twisting, and may be tremulous [4]. The postures are
exaggerated on voluntary actions and during stress and subside
during sleep. It may be painful, if severe and continuous. In
the affected body parts, muscle tone is typically variable,
fluctuating from low to high. Children also demonstrate a
splaying approach (spreading of fingers while approaching an
object) and striatal toe sign (intermittent/persistent extension
of the great toe). Sometimes, patients may show the oscillatory
movement of limbs due to intermittent muscle contractions, known
as dystonic tremors [5]. Often dystonia co-exists with
spasticity in children with a severe brain injury like those
with cerebral palsy.
According to a recent consensus [4], each
patient with dystonia should be classified on a set of clinical
(axis I) and etiological (axis II) descriptors (Fig. 1),
as it aids in diagnosis and treatment. Historically, dystonia
has been classified as primary and secondary. Primary refers to
conditions that manifest with pure dystonia, without any
associated other neurological features and without evidence of
pathological abnormalities. All non-primary dystonia are
labelled as secondary. In this review, disorders are grouped
based on the presence of associated features, with an added
category of acute-onset dystonia and paroxysmal dystonia.
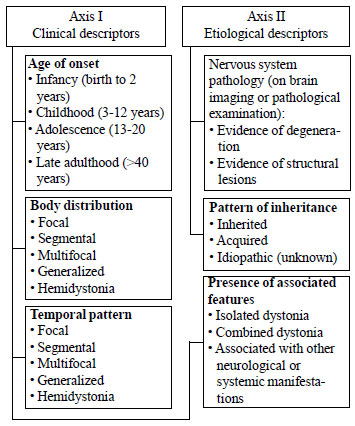 |
|
Fig. 1 Classification of
dystonia [4].
|
Isolated Dystonia
Isolated dystonia refers to conditions in
which dystonia is the only motor feature, with an exception of
tremors [4]. Almost all these conditions are genetic in origin
and most children have a period of normal motor development
before the onset of dystonia. Most often there is a focal onset
of dystonia with gradual progression. As most dystonia are
associated with other movement disorders/neurological
comorbidities; with the increasing number of cases described in
literature, entities with isolated dystonia are shrinking.
DYT-TOR1 dystonia is the most common entity
within this group. It an autosomal dominant disorder with onset
in late childhood or adolescence. It starts with focal limb
dystonia, later progressing to generalized dystonia. DYT-HCPA
dystonia starts within first decade with upper limb and cervical
dystonia. Other forms of isolated dystonia can start with
cranial or cervical dystonia (DYT-THAP and DYT-ANO3) [5,6].
Combined Dystonia
This group includes conditions in which
dystonia is accompanied by features of parkinsonism or
myoclonus, in absence of other neurological abnormalities.
Dopa-respon-sive dystonia or Segawa disease (DYT-GCH1) is an
impor-tant condition in this group. It is an autosomal dominant
disorder that presents between 5-10 years of age with limb
dystonia, with more severe involvement of lower limbs. The most
characteristic feature is diurnal variation with children
typically performing motor activities better in the morning or
after a nap. Few children have some associated features of
parkinsonism. Some cases may mimic spastic cerebral palsy.
Dopa-responsive dystonia is a treatable condition and small to
moderate doses of levodopa bring about a complete response. This
also forms the basis of a trial of levodopa in any child with
dystonia [5,7].
Other entities in this group include
myoclonus-dystonia (DYT-SGCE, DYT-ANO3, DYT-TOR1A or
DYT-CACNA1B) and rapid-onset dystonia-parkin-sonism
(DYT-ATP1A3). Myoclonus-dystonia is genetically heterogeneous
and can present any time after infancy with upper body myoclonus
and limb dystonia. In patients with rapid onset
dystonia-parkinsonism, symptoms are trigge-red with emotional or
physical stress and there is often a stuttering course [5,8].
Associated With Other Manifestations
Children with cerebral palsy, and
neurodegenerative and metabolic disorders form a major part of
this group. Dystonia is often a feature in children with
bilirubin induced neurological damage (BIND) and severe
hypoxic-ischemic brain injury at birth.
Monoamine neurotransmitter disorders are a
hetero-geneous group of conditions that result from deficiency
of cerebral dopamine, serotonin, or both. Most patients become
symptomatic in infancy or early childhood with varying
combination of developmental delay, encephalo-pathy, epilepsy,
spasticity, dystonia, chorea and autonomic dysfunction. Some
children may show diurnal variation. Analysis of CSF
neurotransmitter levels aid in diagnosis. Dopa-responsive
dystonia is also a monoamine neuro-transmitter disorders but has
fewer manifestations [9]
Progressive dystonia and spasticity in early
childhood may be a manifestation of hypomyelinating
leukoence-phalopathies like Pelizaeus Merzbacher syndrome, which
typically presents with pendular nystagmus, developmental delay
and hypotonia in early infancy. Dystonia and ence-phalopathy can
be seen in organic academia and mitochondrial encephalopathies
[5]. Pantothenate kinase–associated neurodegeneration (PKAN)
presents around 3 years of age with clumsiness and gait
abnormality due to lower limb dystonia and spasticity, along
with pigmentary retinopathy [10].
In children with onset of dystonia in late
childhood or adolescence, Wilson disease is an important
differential. Besides dystonia, tremors, dysphagia, dysarthria,
drooling and walking difficulty may be present. Almost all
patients have Kayser-Fleischer rings on eye examination [11].
Acute-Onset Dystonia and Paroxysmal Dystonia
Acute-onset dystonia can be a manifestation
of adverse effects of drugs, stroke, encephalitis, and
functional disorder. Anti-emetics (like metoclopramide) and
antipsychotic drugs (like haloperidol and risperidone) are most
commonly implicated in drug induced dystonia [12]. Dystonia is a
common manifestation of neurotuberculosis and Japanese
encephalitis.
Some genetic conditions manifest with
episodic involuntary movements lasting from seconds to hours,
most often with well-defined triggers. In between the episodes,
the child is usually normal. This group includes conditions like
Paroxysmal kinesigenic dystonia (triggered by sudden movement),
Paroxysmal non-kinesigenic dystonia (triggered by stress,
alcohol, etc.) and Paroxysmal exertional dystonia (triggered by
exercise) [5,13].
Glut-1 deficiency is a rare condition that
occurs due to a deficiency of glucose transporter type 1 in the
brain. It manifests as absence epilepsy, ataxia, developmental
delay and paroxysmal exertional dystonia in early childhood. Low
CSF glucose is the biochemical hallmark and symptoms show a
dramatic response to ketogenic diet [14].
Status Dystonicus
Status dystonicus, or dystonic storm is a
life-threatening condition characterized by frequent or
continuous severe episodes of generalized dystonic spasms.
Although it can occur in any condition causing dystonia, it is
most often seen in children with cerebral palsy and
neurodegenerative disorders. Infections (febrile illnesses) and
other stressors act as triggers. Severe spasm may result in
pain, dehydration, respiratory compromise, rhabdomyolysis and
acute renal failure [15,16].
Clinical Approach
While evaluating a child with dystonia,
ascertain the age of onset, distribution (focal or generalized),
temporal pattern (i.e. diurnal, static, or progressive) and
associated neurological and systemic abnormalities. An important
clue to etiology is that primary dystonia begins as action
dystonia and can persist in kinetic form while secondary or
symptomatic dystonia often begins as sustained postures or tonic
form. Fig. 2 provides an algorithm for clinical
diagnosis.
 |
|
Fig. 2 Clinical approach to
diagnosis of dystonia in children.
|
Eye examination and brain MRI help in
narrowing the differential diagnosis. Eye evaluation should be
focused on KF ring, optic atrophy and retinitis pigmentosa. MRI
is essentially normal in primary dystonia (DYT dystonia). MRI
has diagnostic significance in Wilson disease (T2 hyperintensity
in basal ganglia), PKAN (‘Eye of the Tiger’ sign in globus
pallidus), hypomyelinating disorders (white matter changes) and
Japanese encephalitis (bilateral thalamic involvement) [5].
Metabolic screening is warranted in a child
with encephalopathy. In a child with fluctuating weakness, CSF
analysis will help in ruling out neurotransmitter defect and
Glut-1 deficiency. A therapeutic trial of levodopa is
recommended for all children; although, very few dystonias are
levodopa responsive, and there is a lot of variability in dose
range for children who do respond [17]. In patients with
suspected genetic etiology, dystonia gene panel testing may
provide the definitive diagnosis. With the increasing
availability of whole exome sequencing (WES), the diagnostic
process can be significantly shortened [18].
Management
The drugs used in management of dystonia are
detailed in Table I. In most patients with severe
dystonia, outcomes are unsatisfactory. Neurosurgical procedures
like deep brain stimulation (DBS) and intra-thecal baclofen pump
(ITB) can be used in refractory dystonia. DBS has shown
substantial benefits in children with primary dystonia, whereas
in children with dyskinetic cerebral palsy, mild to moderate
improvement occurs [22].
 |
The management of status dystonicus is
multi-pronged. Patients should be monitored for renal functions,
creatine kinase, blood gas, and urine and/or blood myoglobin
levels. Addressing the precipitant and providing supportive care
(hydration, respiratory support, hemodialysis etc.) is
important. The mainstay of therapy is careful use of sedatives.
Chloral hydrate is recommended as initial therapy (30-100 mg/kg
orally every 3-4 hours). Most patients need the addition of
clonidine (initial dose 3µg/kg 8 hourly, can be increased up to
3-5 µg/kg/hour given as 3-hourly dose. In unresponsive patients,
continuous midazolam infusion may be effective. Besides
sedatives, dystonia specific drugs like trihexyphenidyl,
pimozide and tetrabenazine are also required. In patients with
poor response to drugs, DBS should be offered. The role of ITB
is less clear but may be more effective in patients with
concomitant spasticity [15-16].
TREMORS
Tremors refer to rhythmic, regular
back-and-forth or oscillatory movement of part of the body about
a joint axis [23]. Tremors are classified as resting tremors and
action tremors.
Resting tremors are quite rare in childhood;
however, can be seen in juvenile parkinsonism, Wilson disease,
PKAN, Huntington disease and midbrain lesions [24]. Psychogenic
and dystonic tremors can also be present at rest. In context to
developing countries, infantile tremor syndrome (ITS) is an
important cause. ITS typically manifest in exclusively breastfed
infants of vegetarian mothers, with developmental
delay/regression, anemia, skin hyperpigmentation, and tremors.
Tremors usually start with upper limbs and subside during sleep.
Almost all patients have vitamin B12 deficiency and respond to
supplementation [25].
Action tremors are further classified as
simple kinetic, intention, isometric, task-specific and
postural. Simple kinetic tremor is present during simple limb
movements. It is typically a feature of essential tremor.
Essential tremor affects only the upper limbs and family history
is present in many patients. Usually, the onset is in adulthood
or later, but can sometimes occur in children. A duration of 3
years is required for diagnosis [24]. Functional and
drug-induced tremors can also be simple kinetic. In intention
tremor, the amplitude of tremor increases as the body part is
reaching a visual target. It is characteristically seen in
cerebellar disorders but can also be present in midbrain lesions
[24,26].
Postural tremor is seen when a body part is
held in a position against gravity. Each individual has some
physiological postural tremor, best appreciated in an
outstretched hand. This can be exaggerated by stress, fasting,
illness, strenuous exercise, thyrotoxicosis and drugs like
salbutamol and valproate. Isometric dystonia occurs during
sustained muscle contraction against stationary objects like
while holding a book. Exaggerated physiological tremor and
essential tremor can be isometric. Task-specific dystonia is
related to specific tasks like writing or playing an instrument
[24,26].
Clinical Approach and Management
While evaluating a child with tremors, note
the onset, aggravating and relieving factors, drug history and
family history. Examine for tremors along with muscle tone and
gait pattern. Presence of associated dystonia points towards
conditions like PKAN and Wilson disease. Sudden onset and offset
and marked variation in the semiology favours psychogenic
tremor. Some patients with only dystonia may show tremulous limb
movements, referred as dystonic tremors. Other conditions that
mimic tremors include jitteriness, seizures, myoclonus,
shuddering attacks, and stereotypic movements.
Investigations will depend on the suspected
etiology. Thyroid function should be done for enhanced
physiological tremors. Neuroimaging and other relevant workup
should be done for suspected cerebellar disorder or a
neurodegenerative condition. For ITS, vitamin B12 levels should
be obtained.
Propanolol is recommended in severe cases of
essential tremors and patients with physiological tremors who
have functional or social limitations. Other drugs that are
effective in essential tremors include primidone and
benzodiazepines [24,26].
CHOREA
Chorea refers to involuntary, irregular,
non-repetitive dance-like movements of the body parts that
appear to flow from one muscle group to another without
following any pattern. Children with chorea appear hyperactive
or fidgety. The ability to perform voluntary movements remains
unimpaired. Many grown-up children with chorea transform the
choreiform movement into a voluntary act in order to mask it,
referred to as parakinesia [27]. Patients with chorea have motor
impersistence, which refers to the inability to maintain
sustained postures like keeping tongue protruded or arms
outstretched [28]. Like all movement disorders, chorea also
disappears during sleep.
Chorea with large amplitude, rapid flinging
movements, usually affecting the proximal joints is referred as
ballism [23]. Some patients have slower continuous, involuntary
writhing movements affecting the distal upper extremities,
referred to as athetosis. Athetosis is a distinct movement
disorder; however, it co-exists with chorea and ballism, and
represents a clinical spectrum [23].
Chorea due to a known or presumed genetic
cause is referred as primary chorea. Chorea resulting from
infections, injuries, infiltrative conditions or immune mediated
disorders affecting the brain is called as secondary chorea.
Primary Chorea
Huntington disease: It is an
autosomal dominant disorder that manifest in late adulthood with
chorea, dystonia, psychiatric disturbances, and dementia.
Juvenile Huntington disease is rare and more commonly present
with dystonia, parkinsonism, behavior problems, and cognitive
deterio-ration, rather than chorea [29].
Ataxia-telangiectasia:
Choreoathetosis involving the upper extremities is an early
feature of Ataxia-Telangiectasia, however; it is often mild.
Ataxia that develops by 3-6 years of age is the prominent
manifestation and brings the child to medical attention [30].
Benign hereditary chorea: It is an
autosomal dominant condition with median age of onset of 2.5-3
yrs. The intelligence is normal and the condition tends to
become static after the first decade with improvement in
adulthood. Some patients have accompanying hypothyroidism and
pulmonary disease [31].
Others: In some conditions like
spinocerebellar ataxia type 17, ataxia with oculomotor apraxia
and Friedreich ataxia, chorea may be an early feature, though
ataxia predomi-nantes as the disease progresses. Paroxysmal
movement disorders present with intermittent episodes of chorea
and dystonia [13]. There is a growing list of genetic etiologies
of chorea, with mutation in ADCY5 and PDE10A being
important cause of childhood onset movement disorders [32].
Secondary Chorea
Sydenham chorea: This is the most
common cause of acute-onset chorea in children. It is a late
manifestation of acute rheumatic fever and affects children aged
5-15 years. The chorea mainly involves the upper extremities and
at times there may be wide flinging movements (ballism) [27].
The other manifestations include hypotonia, personality changes,
emotional lability, obsessive-compulsive symptoms and
attention-deficit [33]. Diagnosis is mostly clinical as the
laboratory evidence of recent streptococcal infection is often
lacking.
Other immune-mediated conditions:
Chorea may be the presenting or associated feature in children
with systemic lupus erythematosus associated with
anti-cardiolipin antibodies. Chorea may be a feature of
autoimmune encephalitis, the other manifestations being
seizures, encephalopathy and neuropsychiatric disturbances [34].
Chorea associated with brain injury:
Children with dyskinetic cerebral palsy may have chorea,
besides dystonia. Chorea may be a part of neurological sequelae
after viral encephalitis or stroke.
Drug-induced chorea: Certain drugs like
trihexyphenidyl, levodopa, phenytoin and carbamazepine can
precipitate chorea in a child with other types of movement
disorders or brain injury [27].
Clinical Approach and Management
The history should include perinatal events,
previous infections and associated symptoms. The child should be
examined in a distraction-free environment to note the presence
of chorea and any other movement disorder. Video recording of
movements by parents at home help in characterization of
movements. Child should be examined for motor impersistence,
including inability to keep tongue protruded (darting tongue),
maintain sustained arm grip on examiners fingers (milkmaid’s
grip) and keep upper limbs extended above the head with palms
facing inwards [28]. A clinical approach to diagnosis is
discussed in Fig. 3. Diagnostic work-up depends on the
suspected etiology.
 |
|
Fig. 3 Clinical approach to
diagnosis of chorea in children.
|
Atypical antipsychotics like olanzapine or
risperidone are effective in the management of acute onset
chorea or acute exacerbation. Tetrabenazine and anti-epileptics
like valproic acid and carbamazepine are alternatives. Valproic
acid can be tried in patients who fail to respond to
anti-psychotics. Patients with severe forms of Sydenham chorea
or those unresponsive to antipsychotics may respond to
intravenous immunoglobulin or corticosteroids [35,36]. In
patients with Huntington disease, the preferred drugs are
tetrabenazine, olanzapine, risperidone, and recently,
deutetranenazine [37].
TIC DISORDER
Tics refer to repeated, individually
recognizable, intermittent movements, movement fragments, or
sounds that are almost always briefly suppressible and are
usually associated with awareness of an urge to perform the
movement [23]. The premonitory urge to carry out the movement is
often distressing to older patients. Tics can manifest after 5-6
years of age; however, adolescents are most severely affected.
Tics may affect the social functioning of an individual and
cause embarrassment. Tics are presumed to be genetic because of
the high concordance rate between twins (53%); however, the
genetic loci are not known. Mostly, tic is a primary problem,
rarely it may be part of other neurological disorders [38,39].
Tics are classified based on type (motor or
vocal) and duration. A tic can be simple (like eye blinking,
head jerking, facial grimacing, brief vocalization, throat
clearing, and sniffing) or complex (like obscene gestures or
copropraxia, posturing, echolalia, and coprolalia). A tic
disorder that has been there for less than a year or subsides
within a year is labelled as transient tic disorder. Those
lasting more than a year are called chronic and include Tourette
syndrome and persistent motor or vocal tic disorder [38,39].
Tourette syndrome is defined by presence of
multiple motor tics (at least two distinct ones) and one or more
vocal tics which wax and wane, but have persisted for more than
one year [39]. It is commonly associated with behavioral
disorders like attention-deficit hyperactivity disorder (ADHD),
obsessive-compulsive disorder (OCD), anxiety, mood disorders and
disruptive behavior disorders. About two-third of individuals
meet the criteria of ADHD or OCD at some time during the course
of disease [40]. Persistent motor or vocal tic disorder is
defined as having a single or multiple motor or vocal tics which
may wax and wane but have persisted for more than one year. Tics
may sometimes be part of autism spectrum disorder and
neurodegenerative disorders.
Management
Comprehensive behavioral intervention for
tics (CBIT) is efficacious in reducing tics and is recommended
as the initial treatment. CBIT program consists of habit
reversal therapy, relaxation training, and functional
interventions to address situations that sustain or worsen tics.
If CBIT is ineffective or unavailable, pharmacotherapy can be
used. A variety of drugs are effective including alpha-agonists
like clonidine and guanfacine, anti-psychotics like haloperidol,
pimozide and risperidone and anti-epileptics like topiramate.
Alpha-agonists have lower efficacy than anti-psychotics but are
preferred due to favorable side effect profile. Alpha-agonists
also improve the behavioral comorbidities [41,42].
MYOCLONUS AND STARTLE SYNDROMES
Myoclonus is a sudden, brief, shock-like
involuntary movement of the body. It can involve a single body
part, one half of body or whole body. Myoclonus is mostly
spontaneous, but in some conditions it can be induced by an
action or sensory stimuli like light, sound or touch [1]
Hiccups and sleep starts are considered as
physiological forms of myoclonus. Sleep starts occur during
sleep initiation, manifesting with a sense of falling [1].
Benign neonatal sleep myoclonus and benign myoclonus of early
infancy are viewed as developmental conditions (detailed later)
[47]. Many young children manifest myoclonus during febrile
episodes. Myoclonus can be epileptic as in some epileptic
syndromes (like West syndrome and juvenile myoclonic epilepsy)
and neurodegenerative disorders [1].
Opsoclonus-myoclonus syndrome (OMS) is a rare
immune-mediated condition that manifests acutely or sub-acutely
in toddlers with chaotic multi-directional conjugate eyes
movements (opsoclonus), myoclonus, ataxia, irrita-bility and
sleep disturbance. Approximately half of the patients have
associated neural crest tumors (mostly a neuroblastoma). A
combination of pulse corticosteroids or ACTH and intravenous
immunoglobulins are recommen-ded as the initial treatment.
Surgical tumor resection has no effect on the symptoms in the
majority [43,44].
Startle syndromes are conditions
characterized by exaggerated startle in response to a sound,
movement and touch. Hereditary hyperekplexia is an autosomal
dominant disorder that manifest in infants and young child with
exaggerated startle associated with tonic stiffness of the body,
with repeated falls. A bedside test involves demons-tration of
non-habituating head retraction in response to repeated tapping
of the tip of the nose. With increasing age, the severity
improves; however, it can be precipitated by stress or fatigue.
Most children respond to clonazepam in the dose range of 0.01 -
0.1 mg/kg/day [45,46].
Other conditions associated with exaggerated
startle are other forms of hyperekplexia, post-hypoxic and
post-traumatic encephalopathy, encephalitis, brainstem
dysfunc-tion and neurodegenerative disorders like GM1
ganglio-sidosis and Tay-Sach disease [46].
STEREOTYPIC MOVEMENT DISORDERS
Stereotypies refer to repetitive,
non-functional, patterned movements and/or vocalizations that
can be suppressed by distraction. Simple stereotypies like leg
shaking, hair twirling, body rocking, head banging, and humming
are considered as part of normal behavior. Complex stereotypies
like hand flapping, oro-facial movement and eye-poking, which
interfere with functions or are self-injurious are considered as
abnormal [47]. Complex stereotypies are sometimes seen in
typically developing children, but tend to remain stable or
regress with age. More commonly, they are associated with
conditions like autism spectrum disorders, intellectual
disability, Rett syndrome, Down syndrome, phenylketonuria,
visual or auditory impairment, and acquired brain injury
[47,48]. In children with blindness, stereotypies are seen in
more than two-third of patients, the common ones are body
rocking, repetitive handling of objects, hand and finger
movements, eye pressing and eye poking [49]. Behavior therapy
(habit reversal therapy) is the mainstay of treatment for
stereotypies. Drugs like risperidone and fluoxetine have role in
patients with autism [47,48].
PARKINSONISM
Parkinsonism is a hypokinetic movement
disorder, characterized by presence of resting tremors,
bradykinesia (paucity or slowness of movements), rigidity (lead
pipe type) and postural instability. It can occur in conditions
like Huntington disease and Wilson disease, or can be an adverse
effect of tetrabenzine. Juvenile Parkinson disease is a rare
genetic disorder that manifest with parkinsonism and leg
dystonia [50].
FUNCTIONAL MOVEMENT DISORDERS
It refers to involuntary movements that
result from abnormal mental state or condition, and are
incompatible with recognized neurological and medical
conditions. The common presentations in children are tremors,
dystonia and myoclonus; others being gait disturbances, tics,
chorea and tetany. Many children have identifiable precipitating
factors like school examination, bullying, injury, illness,
sexual abuse of the child or family member, parental discord or
domestic violence, and death of a close relative [51-53]. It is
mostly seen in children above 6-7 years and is more common in
girls.
The diagnosis is suggested by a history of
sudden onset, marked variability of symptoms, and sustained
spon-taneous remissions. Examination often shows symptom
variation, incongruous movements, distraction during spontaneous
speech and behavior, the appearance of symptoms, or worsening
during attention and production or suppression of symptoms on
examiner’s suggestion [51]. In psychogenic tremor, entrainment
or alteration with rhythmic tapping of another body part is
seen. For management, behavior therapy and relaxation techniques
are usually employed. Parental education and counseling are
important [53].
DEVELOPMENTAL AND BENIGN MOVEMENT DISORDERS
These are a group of conditions that manifest
during specific developmental phases of childhood in absence of
associated neurological features. They are considered as
manifestation of subtle modification in the developing brain and
have a favorable outcome [54]. The common disorders are detailed
in Table II.
CONCLUSION
Movement disorders in children comprise of a
heterogeneous group of conditions with diverse etiologies. The
predominant conditions are dystonia, chorea, tics and tremors.
Multiple movement disorders coexist in many conditions and often
create diagnostic confusion. Presence of other neurological and
systemic manifestations helps in narrowing the differential
diagnosis. Neuroimaging and genetic studies enables accurate
diagnosis. The management of these conditions is often
challenging. One should always look for easily treatable
conditions like dopa-responsive dystonia and infantile tremor
syndrome.
Contributors: All authors contibuted to
the manuscript preparation, and final approval.
Competing interests: None stated;
Funding: None.
REFERENCES
1. Delgado MR, Albright AL. Movement
disorders in children: definitions, classifications, and
grading systems. J Child Neurol. 2003;18:S1-8.
2. Fahn S. Classification of movement
disorders. Mov Disord. 2011;26:947-57.
3. Wichmann T. Pathophysiologic Basis of
Movement Disorders. Prog Neurol Surg. 2018;33:13–24.
4. Albanese A, Bhatia K, Bressman SB, et
al. Phenomenology and classification of dystonia: a
consensus update. Mov Disord. 2013;28:863-73.
5. Meijer IA, Pearson TS. The twists of
pediatric dystonia: Phenomenology, classification, and
genetics. Semin Pediatr Neurol. 2018;25:65-74.
6. Zorzi G, Zibordi F, Garavaglia B, et
al. Early onset primary dystonia. Eur J Paediatr Neurol.
2009;13:488-92.
7. Segawa M. Autosomal dominant GTP
cyclohydrolase I (AD GCH 1) deficiency (Segawa disease,
dystonia 5; DYT 5). Chang Gung Med J. 2009;32:1-11.
8. Balint B, Bhatia KP. Isolated and
combined dystonia syndromes - an update on new genes and
their phenotypes. Eur J Neurol. 2015;22:610-617.
9. Kurian MA, Gissen P, Smith M, et al.
The monoamine neurotransmitter disorders: an expanding range
of neurological syndromes. Lancet Neurol. 2011;10:721-33.
10. Hayflick SJ, Kurian MA, Hogarth P.
Neurodegeneration with brain iron accumulation. Handb Clin
Neurol. 2018;147:293-305.
11. Roberts EA, Socha P. Wilson disease
in children. Handb Clin Neurol. 2017;142:141-56.
12. Goraya JS. Acute movement disorders
in children: experience from a developing country. J Child
Neurol. 2015;30:406-11.
13. Waln O, Jankovic J. Paroxysmal
movement disorders. Neurol Clin. 2015;33:137 152.
14. Koch H, Weber YG. The glucose
transporter type 1 (Glut1) syndromes. Epilepsy Behav.
2019;91:90-93.
15. Allen NM, Lin JP, Lynch T, et al.
Status dystonicus: a practice guide. Dev Med Child Neurol.
2014;56:105-112.
16. Lumsden DE, King MD, Allen NM. Status
dystonicus in childhood. Curr Opin Pediatr. 2017;29:674-82.
17. Maas RPPWM, Wassenberg T, Lin JP, et
al. l-Dopa in dystonia: A modern perspective. Neurology.
2017;88:1865-1871.
18. Powis Z, Towne MC, Hagman KDF, et al.
Clinical diagnostic exome sequencing in dystonia: Genetic
testing challenges for complex conditions. Clin Genet.
2020;97:305-11.
19. Luc QN, Querubin J. Clinical
Management of Dystonia in Childhood. Paediatr Drugs.
2017;19:447 61.
20. Fehlings D, Brown L, Harvey A, et al.
Pharmacological and neurosurgical interventions for managing
dystonia in cerebral palsy: a systematic review. Dev Med
Child Neurol. 2018;60:356-66.
21. Hegenbarth MA; American Academy of
Pediatrics Committee on Drugs. Preparing for pediatric
emergencies: drugs to consider. Pediatrics. 2008;121:433-43.
22. Elkaim LM, De Vloo P, Kalia SK, et
al. Deep brain stimulation for childhood dystonia: current
evidence and emerging practice. Expert Rev Neurother.
2018;18:773-84.
23. Sanger TD, Chen D, Fehlings DL, et
al. Definition and classification of hyperkinetic movements
in childhood. Mov Disord. 2010; 25:1538-549.
24. Miskin C, Carvalho KS. Tremors:
Essential Tremor and Beyond. Semin Pediatr Neurol.
2018;25:34 41.
25. Goraya JS, Kaur S. Infantile tremor
syndrome: A review and critical appraisal of its etiology. J
PediatrNeurosci. 2016;11:298 304.
26. Prasad M, Ong MT, Whitehouse WP.
Fifteen minute consultation: tremor in children. Arch Dis
Child Educ Pract Ed. 2014;99:130 134.
27. de Gusmao CM, Waugh JL. Inherited and
acquired choreas. Semin Pediatr Neurol. 2018;25:42 53.
28. Kojoviæ M, Bhatia KP. Bringing order
to higher order motor disorders. J Neurol.
2019;266:797-805.
29. Roos RA. Huntington’s disease: A
clinical review. Orphanet J Rare Dis. 2010;5:40.
30. Pearson TS. More than ataxia:
Hyperkinetic movement disorders in childhood autosomal
recessive ataxia syndromes. Tremor Other Hyperkinet Mov.
2016;6:368.
31. Peall KJ, Kurian MA. Benign
hereditary chorea: An update. Tremor Other Hyperkinet Mov.
2015;5:314.
32. Carecchio M, Mencacci NE. Emerging
monogenic complex hyperkinetic disorders. Curr Neurol
Neurosci Rep. 2017;17:97.
33. Punukollu M, Mushet N, Linney M, et
al. Neuropsychiatric manifestations of Sydenham’s chorea: A
systematic review. Dev Med Child Neurol. 2016;58:16-28.
34. Cardoso F. Autoimmune choreas. J
Neurol Neurosurg Psychiatry. 2017;88:412-17.
35. Walker KG, Wilmshurst JM. An update
on the treatment of Sydenham’s chorea: The evidence for
established and evolving interventions. Ther Adv Neurol
Disord. 2010;3:301-9.
36. Dean SL, Singer HS. Treatment of
sydenham’s chorea: A review of the current evidence. Tremor
Other Hyperkinet Mov. 2017;7:456.
37. Coppen EM, Roos RA. Current
pharmacological approaches to reduce chorea in Huntington’s
disease. Drugs. 2017;77:29-46.
38. Leckman JF. Phenomenology of tics and
natural history of tic disorders. Brain Dev.
2003;25:S24-S28.
39. Fernandez TV, State MW, Pittenger C.
Tourette disorder and other tic disorders. Handb Clin
Neurol. 2018;147:343-54.
40. Hirschtritt ME, Lee PC, Pauls DL, et
al. Lifetime prevalence, age of risk, and genetic
relationships of comorbid psychiatric disorders in Tourette
syndrome. JAMA Psychiatry. 2015;72: 325-33.
41. Pringsheim T, Okun MS, Müller-Vahl K,
et al. Practice guideline recommendations summary: Treatment
of tics in people with Tourette syndrome and chronic tic
disorders. Neurology. 2019;92:896-906.
42. Quezada J, Coffman KA. Current
approaches and new developments in the pharmacological
management of Tourette synd-rome. CNS Drugs. 2018;32:33-45.
43. Desai J, Mitchell WG. Acute
cerebellar ataxia, acute cerebellitis, and
opsoclonus-myoclonus syndrome. J Child Neurol.
2012;27:1482-8.
44. Hero B, Schleiermacher G. Update on
pediatric opsoclonus myoclonus syndrome. Neuropediatrics.
2013;44:324-9.
45. Saini AG, Pandey S. Hyperekplexia and
other startle syndromes. J Neurol Sci. 2020 Sep
15;416:117051.
46. Bakker MJ, van Dijk JG, van den
Maagdenberg AM, et al. Startle syndromes. Lancet Neurol.
2006;5:513-24.
47. Katherine M. Stereotypic Movement
Disorders. Semin Pediatr Neurol. 2018;25:19-24.
48. Barry S, Baird G, Lascelles K, et al.
Neurodevelopmental movement disorders - an update on
childhood motor stereotypies. Dev Med Child Neurol.
2011;53:979-985.
49. Fazzi E, Lanners J, Danova S, et al.
Stereotyped behaviors in blind children. Brain Dev.
1999;21:522-28.
50. Niemann N, Jankovic J. Juvenile
parkinsonism: Differential diagnosis, genetics, and
treatment. Parkinsonism Relat Disord. 2019;67:74-89.
51. Hallett M. Functional (psychogenic)
movement disorders - Clinical presentations. Parkinsonism
Relat Disord. 2016;22: S149-S152.
52. Harris SR. Psychogenic movement
disorders in children and adolescents: an update. Eur J
Pediatr. 2019;178:581-585.
53. Pandey S, Koul A. Psychogenic
movement disorders in adults and children: A clinical and
video profile of 58 Indian patients. Mov Disord Clin Pract.
2017;4:763-67.
54. Bonnet C, Roubertie A, Doummar D, et al. Developmental
and benign movement disorders in childhood. Mov Disord.
2010;25:1317-3.



