|
|
|
Indian Pediatr 2019;56:
792-794 |
 |
Novel Nonsense Mutation in ASXL3 causing
Bainbridge-Ropers Syndrome
|
|
Lingyan Qiao 1,2,
Yusheng Liu3,
Juan Ge1 and Tang Li1,2
From 1Medical Department, Qingdao University; 2Department
of Pediatric Endocrinology and Genetic Metabolic Diseases, Qingdao Women
and Children’s Hospital; and 3Department of Pediatric
Surgery, The Affiliated Hospital of Qingdao University; Qingdao, China.
Correspondence to: Dr Tang Li, Department of Pediatric Endocrinology
and Genetic Metabolic Diseases, Qingdao Women and Children’s Hospital,
Qingdao, China.
Email: [email protected]
Received: January 26, 2019;
Initial review: June 08, 2019;
Accepted: July 20, 2019.
|
|
Background:
Bainbridge-Ropers syndrome is a rare autosomal dominant genetic disorder.
Case characteristics: A 26-day-old neonate presented with feeding
difficulties, excessive sleeping, and hirsutism over forehead and
lumbosacral skin. Outcome: Whole-exome sequencing identified a
novel nonsense mutation. Message: We report a novel
mutation in a Chinese neonate with Bainbridge-Ropers syndrome.
Keywords: Hypersomnia, Mutation, Whole-exome
sequencing.
|
|
B
ainbridge-Ropers syndrome (BRPS, OMIM: 615485),
first identified in 2013, is caused by a loss-of-function mutation in
the ASXL3 gene(OMIM: 615115). The clinical features of the
condition are severe psychomotor retardation, speech disorders, feeding
difficulties, hypotonia, and distinctive craniofacial features.
To date, almost all ASXL3 gene variants
reported in the literature are nonsense mutations and frameshift
mutations, except for one splice site mutation (c.3039+1G>A) [1]. We
report a novel nonsense mutation in ASXL3.
Case Report
The patient was a 26-day-old female neonate with
failure to thrive, excessive sleeping and hypotonia. She was the only
daughter of a healthy, non-consanguineous Chinese couple. Mother’s
pregnancy was normal with spontaneous birth in the 39th week of
gestation. She suffered from embryonic developmental arrest at day 58
during the first pregnancy, and spontaneous abortion during the second
pregnancy. The birth weight was 2.6 kg and length was 50 cm. The patient
was hypersomnic and breastfeeding frequency was low. She had a loud cry,
but used to sleep around 22 hours per day. Her weight at presentation
was 2.51 kg (<3 rd
percentile), length was 51 cm (3rd-10th
percentile), and head circumference was 34 cm (3rd
percentile). She had slightly wide and flat nose bridge, hirsute
forehead and lumbosacral skin, and hypotonia. Rooting reflex, sucking
reflex, Moro reflex, grasp reflex, and bilateral knee reflex were
normal. Liver function tests blood gas analysis, blood ammonia, lactate
levels, blood tandem mass spectrometry and urinary organic acid analyses
were normal. Her brain and lumbo-sacral magnetic resonance imaging
showed no gross abnormalities. The result of karyotype analysis were 46,
XX.
At the age of 6 months, her weight was 4.6 kg (<3 rd
percentile), length 63.5 cm (3rd-10th
percentile), and OFC head circumference 40.6 cm (3rd-10th
percentile). Her developmental milestones were
delayed. She barely had steady head control, was not able to rollover or
sitting up unaided. She had little facial expression, and barely made
eye contact with people around (even with parents). However, she could
follow sounds and objects, and had not suffered from any seizures. She
maintained a daily sleep time of 20-22 hours. Craniofacial features
gradually became more prominent (Fig. 1).
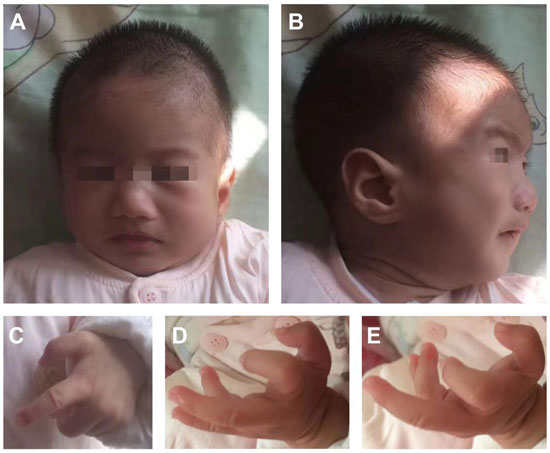 |
|
Fig 1. Facial phenotypes and hands of
patient at age 6 months: (a) long face, arched eyebrows, wide
nose bridge, and downturned corners of the mouth; (c-e)
prominent forehead, short nose bridge, and high-arched palate;
her fingers and thumbs were in bent position and the phalangeal
joints were stiff, with ulnar deviation of both wrists.
|
Whole-exome sequencing revealed a heterozygous
mutation c.3464C>A in exon 12 of ASXL3 gene, resulting in the
amino acid change p.S1155X. No variation was present at this site in her
parents. Sanger sequencing of family members validated this analysis,
suggesting a de novo mutation (Web Fig. 1).
The predicted results of both SIFT and PolyPhen-2 were unknown.
Discussion
In addition to the typical symptoms of craniofacial
features, hypotonia, and ulnar deviation of both wrists, there were
other manifestations (hypersomnia, hirsutism) that have not been
reported in BRPS cases available in the literature. The decreased
feeding frequency was probably related to longer sleep time. Due to
limited knowledge of the disease and lack of specific features in early
infancy, early diagnosis of this syndrome is very challenging. In our
case, the variant is predicted to cause a stop-gain at amino acid 1155
(NM_030632;exon12: c.3464C>A, p.S1155X), resulting in truncate ~50% of
the encoded protein. According to the 2015 ACMG Guidelines [2], the
c.3464C>A mutation was defined to be pathogenic.
The clinical phenotype of BRPS is complex, and
differs even in patients with the same gene variant [3,4]. We speculate
that the clinical heterogeneity of patients with BRPS may relate to the
following two factors. Firstly, it is likely that ASXL3 gene
mutation occurs after fertilization or during early embryonic
development, resulting in the formation of chimeras with different
presentations or incomplete phenotypes [5]. Secondly, the expression of
the ASXL3 gene varies in different tissues, higher in testis,
ovary, and brain tissue. Therefore, the probability of ASXL3 gene
mutation may increase significantly during fertilization and embryonic
brain development.
Bainbridge-Ropers syndrome has an autosomal dominant
inheritance pattern. Kuechler, et al. [6] reported that the elder
twin sister of a patient were healthy and did not carry a mutation in
the ASXL3 gene, indicating that the ASXL3 gene mutation in
that patient was a de novo mutation. On the other hand, the twin
sisters described by Koboldt, et al. [7] were both diagnosed with
Bainbridge-Ropers, and both had the same gene mutation, suggesting
possible germline mosaicism in one parent. In this case, the patient’s
mother had been pregnant three times, with embryonic arrest occurring on
day 58 of the first pregnancy, spontaneous abortion ending the second
pregnancy during the first trimester, and the third pregnancy resulting
in the normal birth of the patient. We speculate that the mutation in
ASXL3 gene might have played a role in the early loss of the first
two pregnancies. In addition, the currently reported pathogenic mutation
of the ASXL3 gene is generally a de novo mutation, which
includes the possibility of a germline chimera. Early genetic counseling
should be performed for families desiring another child to avoid the
birth of more children with the same disease.
Contributors: LQ: Case management and
drafted the manuscript; YL and JG: Literature review and helped to draft
the manuscript; TL: organized the clinical follow-up study and reviewed
the manuscript.
Funding: None; Competing interests:
None stated.
References
1. Hori I, Miya F, Ohashi K, Negishi Y, Hattori A,
Ando N, et al. Novel splicing mutation in the ASXL3 gene causing
Bainbridge-Ropers syndrome. Am J Med Genet A. 2016;170:1863-7.
2. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster
J, et al. ACMG Laboratory Quality Assurance Committee. Standards
and Guidelines for the Interpretation of Sequence Variants: A Joint
Consensus Recommendation of the American College of Medical Genetics and
Genomics and the Association for Molecular Pathology. Genet Med.
2015;17:405-24.
3. Srivastava A, Ritesh KC, Tsan, YC, Liao, R, Su, F,
Cao, X, et al. De novo dominant ASXL3 mutations alter H2A
deubiquitination and transcription in Bainbridge-Ropers syndrome. Hum
Mol Genet, 2016;25;597-608.
4. Balasubramanian M, Willoughby J, Fry AE, Weber A,
Firth HV, Deshpande C, et al. Delineating the phenotypic spectrum
of Bainbridge-Ropers syndrome: 12 new patients with de novo,
heterozygous, loss-of-function mutations in ASXL3 and review of
published literature. J Med Genet. 2017;54:537-43.
5. Bainbridge MN, Hu H, Muzny DM, et al. De
novo truncating mutations in ASXL3 are associated with a novel clinical
phenotype with similarities to Bohring-Optiz syndrome. Genome Med.
2013;2:11.
6. Kuechler A, Czeschik JC, Graf E, Grasshoff U,
Hüffmeier U, et al. Bainbridge-Ropers syndrome caused by
loss-of-function variants in ASXL3: A recognizable condition. Eur J Hum
Genet. 2016;25:183-91.
7. Koboldt DC, Mihalic Mosher T, Kelly BJ, Sites E,
Bartholomew D, Hickey SE, et al. A de novo nonsense mutation in ASXL3 shared
by siblings with Bainbridge-Ropers syndrome. Cold Spring Harb Mol Case
Stud. 2018;4:a002410.
|
|
|
 |
|
