|
|
|
Indian Pediatr 2019;56:
789-791 |
 |
Chinese Siblings with Prader-Willi Syndrome Inherited from
Their Paternal Grandmother
|
|
Dai Yang-Li 1,
Huang Ke1, Zou
Chao-Chun2 and
Dong Guan-Ping1
From Departments of 1Endocrinology and 2Child
Healthcare, Children’s Hospital Zhejiang University School of Medicine,
Zhejiang, China.
Correspondence to: Dr Zou Chao-Chun,
Department of Child Healthcare, Children’s Hospital Zhejiang University
School of Medicine, 3333 Binsheng Road, Hangzhou 310052, China.
Email: [email protected]
Received: December 20, 2018;
Initial review: May 20, 2019;
Accepted: July 20, 2019.
|
|
Background: Prader-Willi syndrome
(PWS) is a complex neurobehavioral disorder caused by failure of
expression of paternally inherited genes in the PWS region of
chromosome 15. Case characteristics: Two siblings who both met
the inclusion criteria for clinical diagnosis of PWS during neonatal
period. Outcome: Molecular genetic analysis demonstrated a 417-kb
microdeletion within the 15q11.2 region inherited from siblings’
paternal grandmother, involving key genes of PWS, except for
UBE3A, which may explain why their father and paternal grandmother
had a normal phenotype. Conclusion: The findings may be helpful
for better understanding of the underlying mechanism of this rare
imprinting defect.
Keywords: Microdeletion, Mode of inheritance,
Molecular genetic analysis.
|
|
P
rader-Willi syndrome (PWS; MIM 176270) is a
genomic imprinting disorder – 70% of individuals with PWS have a de
novo paternal microdeletion of the chromosome 15q11.2-q13 region
(so-called PWS/AS region), namely paternal deletion type. About 25% of
individuals have maternal disomy 15 of chromosome 15q11.2-q13 region,
namely uniparental disomy (UPD) type. Less than 3% have defects in the
genomic imprinting due to microdeletion or epimutation [1]. On very rare
occasions, chromosomal translocations or rearrangements of the
15q11.2-q13 region have been reported [2]. The epigenetic and genomic
changes causing PWS entirely lead to a loss of the expression of the
paternally expressed genes on 15q11.2-q13 region because the maternal
contribution for these genes has been programmed by epigenetic factors
to be silenced [3]. Conversely, loss of the expression of preferentially
maternally expressed UBE3A in this region by several possible
mechanisms may lead to Angelman syndrome [4]. We report two siblings
with PWS due to shorter microdeletions inherited from their paternal
grandmother.
Case Report
The proband was a Chinese 20-day-old male infant born
to non-consanguineous parents (parents’ age 28 years) at gestational age
of 40 weeks by caesarean section. During pregnancy, mother perceived
decreased intrauterine movement. He had birth weight of 3.15 kg and
length of 50 cm. Hypotonia, weak cry, weak suck, and feeding problems
were noted after birth. Physical examination showed lethargy, abnormal
facies (narrow face, micrognathia, high-arched palate, a thin upper lip
with a down-turned mouth), generalized hypopigmen-tation, brown hairs,
small genitalia, cryptorchidism, and poor reflexes. Biochemical
analyses, including blood electrolytes, liver and renal function tests,
thyroid function test, and serological testing for perinatal infections
were normal. The parents were healthy, with normal intelligence, except
that the mother had hepatitis B virus (HBV) infection with normal liver
function.
His older sister was vaginally delivered at 41 weeks
of gestation, and had died at 4 day of age. She was hypotonic, never
cried, and had a weak suck. She also had lethargy, abnormal facies (micrognathia,
a thin upper lip with a down-turned mouth), brown hairs, external
genital deformity, strephexopodia, and poor reflexes. The pedigree of
the proband’s family and pictures of the siblings are shown in Fig.
1 and 2.
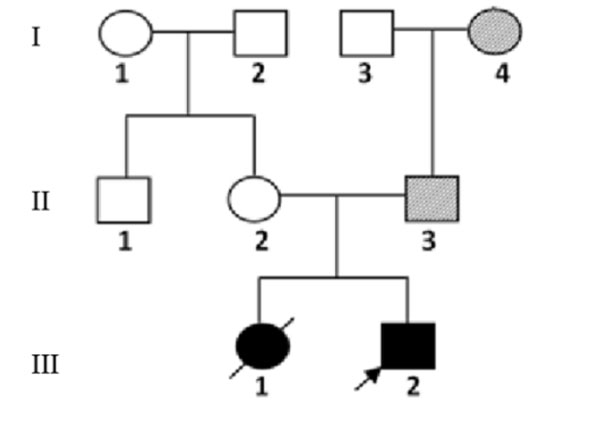 |
|
Fig. 1 The three-generation pedigree
of the affected family.
|
 |
|
Fig. 2 The clinical pictures of (a)
proband, and (b) his sister. Both showed abnormal facies (narrow
face, micrognathia, almond eye, a thin upper lip with a
down-turned mouth), generalized hypopigmentation, and brown
hairs.
|
As two siblings were affected, hereditary disease was
suspected. Karyotyping (450-550 bands) was done for the proband, his
sister, and his parents in a local hospital by Guangzhou Kingmed
Diagnostics Group Co. Ltd. (Guangzhou, China). A total of 50 metaphase
cells were analyzed. The karyotypes of the proband, and his father were
normal, while the karyotype of his mother was 46, XX 9qh+.
Single nucleotide polymorphism (SNP) based
chromosomal microarray analysis was performed by using the Infinium
OmniZhongHua-8 kit (San Diego, CA, USA) according to the manufacturer’s
instructions. The KaryoStudio software (Illumina, Inc., San Diego, CA,
USA), and Database of Genomic Variants (DGV) were utilized to detect and
analyze copy number variations (CNVs). A 417-kb microdeletion in
chromosome 15q11.2 [Hg19 arr15q11.2(24 963 375-25 380 656)×1] was found
for the proband and his father. This region involved the imprinting
center of PWS/AS, SNURF-SNRPN and SNORD107, SNORD64, SNORD 109A,
SNORD116, SNORD115, and SNORD109B. A 206-kb microdeletion in chromosome
4p16.1 [arr4p16.1(8 218 420-8 424 831)×1] and a 149-kb microduplication
in chromosome 16p13.2 [arr16p13.2(8 798 528-8 948 473)×3] were found for
his mother.
The analysis was subsequently followed by methylation
sensitive multiplex ligation-dependent probe amplification (MS-MLPA) of
the proband, his father, and paternal grandmother to confirm the
diagnosis of PWS; and explore the origin of variations using SALSA
MS-MLPA ME028-B2 Prader Willi/Angelman kit (MRC Holland, Amsterdam, The
Netherlands). This kit contains 46 probes, 32 of which are specific for
sequences in or close to the PWS/AS critical region on 15q11.2-q13 which
can be used to detect CNVs in this region. A heterozygous microdeletion
containing the imprinting center ranging from SNRPN intron u1 through
the SNRPN locus and SNORD 107 to SNORD 109B snoRNA cluster located at
15q11.2 was found on the proband, his father, and his grandmother. DNA
methylation abnormalities were noted as well, including 100% methylation
for the proband, and 0% methylation for his father and grandmother.
The proband, his father, and his grandmother all
underwent linkage analysis, and the analysis showed that the alleles of
SNRPN-12N, -13N, -14N, and -15N were dropped out.
These allelic sites were used for the preimplantation
genetic diagnosis (PGD) based on linkage analysis. It was revealed that
5 of the 12 blastocysts were normal and the mother was ready to receive
the implantation.
Discussion
PWS is an imprinted neurobehavioral condition,
influencing several organs, and mainly occurs due to the absence of
expression of a cluster of paternally expressed genes located at
15q11-q13. PWS is usually sporadic; however, in some families, an
epimutation or incomplete processing of imprinting in germ cell may
negatively influence the process, originating from father or from
microdeletion of the DNA imprinting center. The mentioned microdeletion
defect has been reported in about 15% of individuals with PWS due to
imprinting defect (ID) [5]; although, recent studies have indicated a
possible higher rate of microdeletion in the imprinting center (IC) [6].
This microdeletion can be derived from the paternal grandmother through
the father, and may lead to birth of another child with PWS, and its
risk may reach 50%. However, if microdeletion only passes through the
maternal line, no phenotypic effect may occur, while her sons may be at
a 50% risk of having children with PWS, and her daughters’ sons may also
be at risk of having children with PWS [7]. In the majority of reported
cases, the IC deletion was familial, and in contrast, none of the
patients with a non-IC deletion have an affected sibling [8].
The proband and his older sister both met major and
minor criteria for clinical diagnosis of PWS presented by Holm, et al.
[9,10], and the diagnostic scores were both equal to 5.5. Hartin, et
al. [7] reported the clinical and genetic findings in three adult
siblings with PWS caused by a microdeletion in the chromosome 15
imprinting center inherited from an unaffected father that controls the
activity of genes in the 15q11-q13 region. They demonstrated that the
PWS siblings’ mother displayed a normal copy number for the same probes,
indicating that she did not carry a microdeletion in the PWS imprinting
center [10]. The maternal imprint could not be erased in the paternal
germline in those PWS cases with imprinting center defects.
In conclusion, the wide variety of phenotypes that
involve multiple organ systems match with the genetic complexity of the
PWS chromosomal region, with multiple imprinted genes, gene duplication
and CNVs, alternative splice variants, and the mechanisms of imprinting.
Reporting of families with more than one affected individuals with PWS,
which were not related to translocation or inversion of chromosome 15,
can be helpful to identify the genetic defect within the imprinting
center.
Contributors: Dai YL, Huang K: diagnosed and
managed the case, and drafted the initial manuscript; Dong GP, Zou CC
supervised the patient care and reviewed and revised the manuscript. All
authors approved the final manuscript and agree to be accountable for
all aspects of the work.
Funding: This work is supported, in part, by
grants from the National Natural Science Foundation of China (Grant No.
81170787, 81371215, 81670786), and the Zhejiang Provincial Program for
the Cultivation of High-Level Innovative Health Talents (2014).
Competing Interest: None stated.
References
1. Butler MG. Prader-Willi syndrome: Obesity due to
genomic imprinting. Curr Genomics. 2011;12:204-15.
2. Rocha CF, Paiva CL. Prader-Willi-like phenotypes:
A systematic review of their chromosomal abnormalities. Genet Mol Res.
2014;1:2290-8.
3. Cassidy SB, Driscoll DJ. Prader-Willi syndrome.
Eur J Hum Genet. 2009;17:3-13.
4. Williams CA, Driscoll DJ, Dagli AI. Clinical and
genetic aspects of Angelman syndrome. Genet Med. 2010;12: 385-95.
5. Buiting K, Gross S, Lich C, Gillessen-Kaesbach G,
El-Maarri O, Horsthemke B. Epimutations in Prader-Willi and Angelman
syndromes: A molecular study of 136 patients with an imprinting defect.
Am J Hum Genet. 2003;72: 571-7.
6. Newkirk HL, Bittel DC, Butler MG. Analysis of the
Prader-Willi syndrome chromosome region using quantitative microsphere
hybridization (QMH) array. Am J Med Genet Part A. 2008;146A:2346-54.
7. Hartin SN, Hossain WA, Weisensel N, Butler MG.
Three siblings with Prader-Willi syndrome caused by imprinting center
microdeletions and review. Am J Med Genet Part A. 2018;176:886-95.
8. Buiting K, Gross S, Lich C, Gillessen-Kaesbach G,
Maarri O, Horsthemke B. Epimutations in Prader-Willi and Angel-man
Syndromes: A Molecular study of 136 patients with an imprinting defect.
Am J Hum Genet. 2003;72:571-7.
9. Holm VA, Cassidy SB, Butler MG, Hanchett JM,
Greenswag LR, Whitman BY, et al. Prader-Willi syndrome: Consensus
diagnostic criteria. Pediatrics. 1993;91:398-402.
10. Gunay-Aygun M, Schwartz S, Heeger S, O’Riordan
MA, Cassidy SB. The changing purpose of Prader-Willi syndrome clinical
diagnostic criteria and proposed revised criteria. Pediatrics.
2001;108:E92.
|
|
|
 |
|
