|
|
|
Indian Pediatr 2019;56: 757-766 |
 |
Testing Modalities for
Inborn Errors of Metabolism – What a Clinician Needs to Know?
|
|
Sunita Bijarnia-Mahay 1
and Seema Kapoor2
From 1Institute of Medical Genetics and
Genomics, Sir Ganga Ram Hospital; and 2Division of Genetics,
Department of Pediatrics, Maulana Azad Medical College and associated
Lok Nayak Hospital; New Delhi, India.
Correspondence to: Dr Sunita Bijarnia-Mahay,
Institute of Medical Genetics and Genomics, Sir Ganga Ram Hospital,
Rajinder Nagar, New Delhi 110 060, India.
Email: [email protected]
|
The present century is being hailed as the century
for genetic therapies, and inborn errors of metabolism is leading the
way. As we gear ourselves for treating children with genetic and
metabolic disorders, the key is to recognize them early and accurately
for best outcomes. In these changing times with advent of technology,
clinicians are now more aware, exposed and well equipped with the
armamentarium of diagnostic modalities. However, it is difficult to
choose between the tests without a baseline knowledge about testing for
genetic and metabolic disorders. The key question for a clinician when
dealing with a suspected metabolic disorder case is ‘what test to order’
and ‘how to proceed.’ The current article provides a rational view on
the various laboratory testing modalities available for diagnosis of
inborn errors of metabolism. The article provides details of the basic
and advanced metabolic tests that can be ordered in appropriate
settings.
Keywords: Diagnosis, Genetics, Metabolic disorders, Tandem
mass spectrometry.
|
|
A
s we begin to settle down in the 21st
century, the realization that technology has made it possible to move
its application from the bench to the bedside is dawning. Improvements
in neonatal health care have led to an unprecedented decline in infant
mortality rate to 33 per thousand live births [1], shifting the focus on
to other ‘significant’ causes of pediatric morbidity and mortality.
Inborn errors of metabolism (IEMs), the term coined by Sir Archibald
Garrod in 1908 [2], are a group of disorders which affect nearly 1 in
1000 children [3]. The diagnostic ability has preceded the therapeutic
advances by a few decades. The recognition of inherited metabolic
disorders has increased with availability newborn screening and
metabolic testing in both public and private domain. Technological
advances in the field of diagnostics has benefitted patients with
metabolic disorders and this appears to be appropriate time to focus on
the correct technology to use [4,5]. This review will elaborate upon the
various testing modalities for screening and confirmative diagnosis of
the IEMs.
A common presentation of IEMs is an apparently
healthy child presenting acutely with a short history of illness, which
is worsening progressively. The symptoms could be as mild as a fever, or
as severe as encephalopathy or status epilepticus. Few common clinical
presentations are (i) acute (and recurrent) episodes of symptoms
such as vomiting, respiratory distress, ataxia, lethargy or coma with or
without seizures, hypotonia, hypoglycemia or hyperammonemia; (ii)
chronic and progressive symptoms such as developmental delay, epilepsy
or other neurological illness, or (iii) organ specific
presentation (single or multiple) such as hepatopathy, cardiomyopathy,
neuro-muscular, renal, gastroenterology or hematological illness. These
children should be evaluated for a metabolic cause particularly with a
past history or family history of similar illness in a sibling. Presence
of consanguinity makes the suspicion of an IEM stronger. A complete
discussion on the various clinical approaches to diagnosis of IEMs is
out of scope of this review; the readers are suggested to refer to other
resources [6-8].
IEMs can be detected on routine tests such as blood
gas, blood glucose, blood ammonia and ketones in urine, which can be
followed by more advanced tests. The tests utilized for diagnosing an
IEM are detailed below.
Basic Metabolic Tests
Some tests on blood and urine can be performed at the
bedside or quickly in a routine laboratory. These tests are helpful in
screening for many IEMs, but are not diagnostic by themselves.
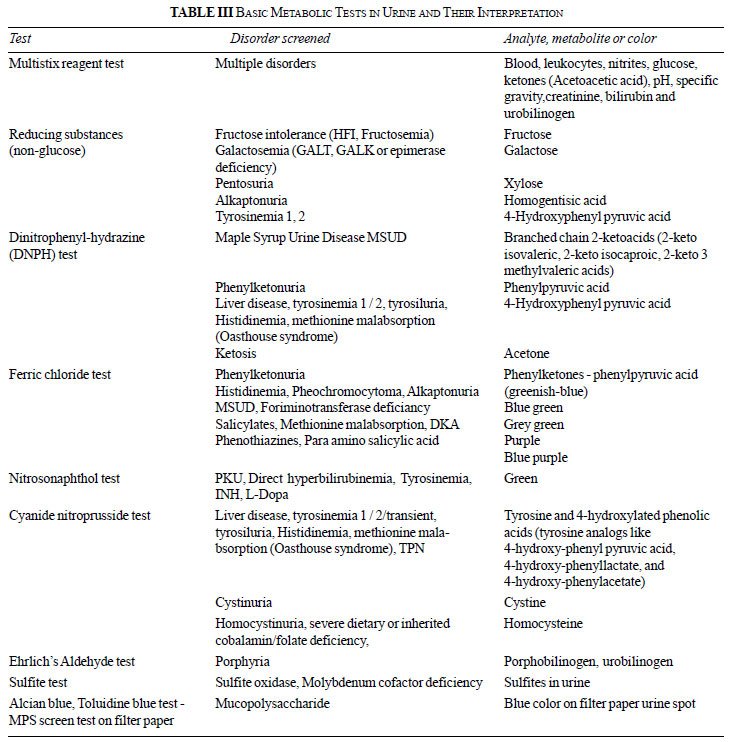
Urine Analysis
The urine is an excellent source of crucial
metabolites as the excess pathological metabolites in the body are
excreted out in urine. Urine should be examined for the following
observations before subjecting it to advanced biochemical testing
[8-12].
Color: Yellow (light to dark) may indicate
bilirubin/drug metabolites, while coffee or cola colored may indicate
rhabdomyolysis, and darkening on standing (exposure to light) may be
observed in porphyria and alkaptonuria (Table I).
TABLE I Interpreting Abnormal Color and Odour of Urine
|
Characteristic |
Disorder |
Compound |
|
Urine Color |
|
Dark brown or black
|
Alkaptonuria |
Homogentisic acid |
|
Hemoglobinuria/Myoglobinuria |
Hemoglobin/Myoglobin |
|
Red
|
Hematuria |
Erythrocytes |
|
Porphyrias |
Uroporphyrin, Coproporphyrin |
|
Ingestion of colored foods - beet |
Anthrocyanine |
|
Red dyes |
Rhodamine B, Phenolphthalein |
|
Blue |
Hartnup disease |
Indican |
|
Blue/brown |
Alkaptonuria |
Homogentisic acid |
|
Urine Odor |
|
Maple syrup/ burnt sugar |
Maple syrup urine disease |
Sotolone, 2-oxoisocaproic acid |
|
Sweaty feet |
Isovaleric acidemia, Glutaric acidemia type II |
Isovaleric acid |
|
Sulfur |
Cystinuria |
Hydrogen sulfide |
|
Boiled cabbage |
Tyrosinemia type I, Methionine malabsorption |
2-hydroxybutyric acid, 2-keto-4-methiolbutryric acid |
|
Old fish |
Trimethylaminuria, Dimethylglycine
|
Trimethylamine, dimethylglycine |
|
dehydrogenase deficiency |
|
|
Cat’s urine |
Multiple carboxylase deficiency,
|
|
|
3-methyl crotonyl-CoA carboxylase deficiency |
3-Hydroxyisovaleric acid
|
|
Mousy |
Phenylketonuria |
Phenylacetic acid |
|
Maple syrup/ burnt sugar |
Maple syrup urine disease |
Sotolone, 2-oxoisocaproic acid |
Odor: Sweet odor in ketosis, maple syrup/ burnt
sugar in maple syrup urine disease (MSUD) and mousy in phenylketonuria
(PKU) have been described. Table I outlines the odors
described in the various IEMs.
Urinalysis: Urinary pH, specific gravity,
creatinine concentration, glucose/reducing substances,
protein/albumin/micro-albumin, ketonuria are important preliminary
investigations in IEMs.
Blood Tests
Complete blood count with differential counts:
Anemia, thrombocytopenia, leucopenia or leucocytosis is noted in organic
acidurias. Peripheral blood smear is helpful for the type of anemia,
signs of hemolysis, and for abnormal cells. Megaloblastic anemia can
point towards a vitamin B 12/cobalamin
or folate absorptive or intra-cellular utilization disorder.
Blood Glucose, Electrolytes, Lactate, Acid base
balance/arterial blood gas, Ammonia, and Ketosis (Acronym- GELAK):
Stringent conditions need to be met with regards to the transport and
analysis of both lactate and ammonia. Ammonia estimation should
be done from a free flowing blood sample obtained from either arterial
or venous puncture. The sample should be collected in a preferably
pre-chilled tube containing EDTA (for ammonia) as an anticoagulant.
Patient should be fasting (or at least 4-6 hours after feeding) and
non-stressed such that it is not collected after a difficult
venipuncture. This, however, would not hold true for an acutely sick
child when samples should be collected immediately upon arrival or in
acutely decompensated state, irrespective of feeding status. Samples
should be transported on ice, separated within 15 minutes of collection,
and analyzed immediately to prevent artefactual increase in generation
of ammonia from RBC degradation or deamination of amino acids by enzymes
such as gamma glutamyl transferase. Normal levels of ammonia are <110
µmol/L (neonates), <80 µmol/L (infants), <50 µmol/L (older children) and
<35 µmol/L (adults). Lactate analysis can be done from whole blood or
plasma collected in a sodium fluoride or heparinized vial. Sample should
be transported on ice slurry or an ice pack within 30 minutes of
collection. Once aliquoted, the plasma is stable on room temperature for
8 hours. Blood collected from artery is ideal but a free flowing venous
blood sample is also acceptable. The lactate value on blood gas is
reliable if the machine is calibrated. There are two units used
frequently for lactate values – mg/dL and mmol/L. Normal range of
lactate (both in blood in CSF) are 2-20 mg/dL and 0.2–2.0 mmol/L at all
ages.
Liver function tests (LFT): The list of IEMs with
liver involvement is exhaustive–ranging from small molecule disorders
such as urea cycle, amino acid or organic acid disorders to
mitochondriopathies and large molecule disorders such as peroxisomal and
lysosomal storage disorders. For more in-depth information on IEMs
related to liver disease readers are directed towards other reviews
[13,14].
Renal function tests: Serum creatinine indicates
renal function derangement, if high; and creatine synthetic or
transporter defect, if low.
Uric acid: High levels in presence of features
such as intellectual disability and self-mutilation is highly suggestive
of Lesch-Nyhan Syndrome (HPRT deficiency – purine recycling disorder).
High levels may also suggest disorders of carbohydrate disorders such as
glycogenolysis or gluconeogenesis. A low level may point towards
xanthine/hypoxanthine disorder or a molybdenum cofactor deficiency (MoCD).
Creatine kinase: It is elevated in metabolic
disorders affecting the muscle causing glycogenolysis, or rhabdomyolysis,
and in disorders of energy production such as fatty acid oxidation
defects, gluconeogenesis defects or a mitochondrial disorder.
Plasma total homocysteine level: This is elevated
in disorders of vitamin B 12
or folate metabolism, and in classic homocystinuria due to deficiency of
cystathionine beta-synthase enzyme that utilizes pyridoxine as a
co-factor. Low homocysteine level may be indicative of a methionine
disorder such as methyl adenosyl transferase deficiency. Normal range
for plasma homocysteine is 5-15 µmol/L at all ages.
Lipid profile: This is deranged in glycogen
storage disorders, lipoprotein disorders, transient infantile
hypertriglyceridemia. Dyslipidemia is also noted in Wolman disease,
fructose-1-6-bisphosphatase deficiency and lipid storage disorders such
as Niemann-Pick disease.
Prolactin levels: This may be elevated in
neurotransmitter disorders (dopamine synthesis). Normal level in serum
is 5 to 20 ng/mL (5 to 20 µg/L).
Copper levels (in plasma): This may be
decreased in Wilson disease, Menkes, aceruloplasminemia, and MEDNIK
syndrome; and increased in peroxisomal disorders.
Iron levels: Increased serum iron/ferritin is
observed in hemochromatosis and peroxisomal disorders
Advanced Metabolic Tests
These are biochemical reactions utilized for making a
specific diagnosis of a single or group of disorders. These tests
measure specific metabolite, enzyme or a cofactor. Specific indications
for these tests with classification of IEMs are provided in Table
II. The classification is proposed by the Society for study of
inborn errors of metabolism (SSIEM) according to biochemical basis [7].
TABLE II Metabolic Tests for Diagnosis of IEMs (According to SSIEM Classification ) [7]
|
Disease group/disorder |
Metabolic derangements* |
Basic metabolic tests for diagnosis |
Diagnostic modalities of choice |
|
Disorders of amino acid,organic acids and peptidemetabolism |
Acidosis, hypoglycemia,hepatic and renal dysfunction,
cardiomyopathy |
GELAK (glucose, electrolytes,lactate, acid-base, ammonia,ketones),
homocysteine |
Tandem Mass spectrometry (MS/MS), U/HPLC
quantitative amino acids, Urine GC-MS, Succinylacetone (Tyrosinemia
type 1) |
|
Disorders of carbo-hydrate metabolismDisorders of fatty acidand
ketone body meta-bolism |
Hypoglycemia, ketosis, acidosis, hepatic and renal
dysfunctionHypoglycemia, rhabdo-myolysis,hyperammonemia,
cardio-myopathy, liver dysfunction,Reye-like syndrome |
Glucose, other sugars, insulin, acid-base, lactate, ketones,
LFTGlucose, lactate, ammonia,ketones, acid-base, Creatine kinase,
uric acid
|
Fasting studies, enzyme assays (Galactosemia), genetic
testingMS/MS, Urine GC-MS
|
|
Disorders of energymetabolism |
Recurrent acidosis, hyperlactatemia, hyperammonemia,liver
dysfunction, Myopathy, cardiomyopathy, renal tubular dysfunction |
Glucose, lactate, Pyruvate, Kreb cycle metabolites, ketones,organic
acids |
Lactate/ pyruvate ratios (plasma, CSF), Urine GC-MS,Mitochondrial
respiratory chainenzymology, mtDNA and nDNA gene studies |
|
Disorders in the meta-bolism of purines , pyrimidines and
|
Increased or decreased uric acidand xanthinesReduced
cholesterol, deranged |
Uric acid in serum, urinecrystals, urine xanthine/hypoxanthine |
Uric acid in serum, urine. Purine and pyrimidines in urine
by HPLC, gene studies nucleotides |
|
Disorders of sterol and bile acid synthesis |
liver enzymes, cholestasis,deranged coagulation profile |
Cholesterol in serum , lipid profile, liver function tests,
coagulation factors, Vitaminlevels (A, D, E, K) |
Sterol assays in plasma: 7- dehydrocholesterol etc for bile acid
synthetic defect – specific bile acids by FAB-MS, gene studies |
|
Disorders of porphyrinand haem metabolism |
Increased porphyrins in blood and urine |
Porphyrins in blood and urine |
Screening for Porphobilinogen in urine (Hoesch test,
Watson-Schwartz test) qualitative/quantitative Specific
porphyrins in urine, stool or blood. Gene studies |
|
Disorders of lipid and lipoprotein metabolism |
Abnormal lipid profile, insulin |
Lipid profile, insulin, liver enzymes |
Enzyme assays, gene studies |
|
Congenital disorders of glycosylation and other disorders of
proteinmodification
|
Abnormal Liver function, insulin, coagulation factors
|
Liver function test and coagulation profile |
Transferrin isoforms pattern by CZE, IEF or other
method like HPLC. Enzyme assay(skin fibroblasts), gene studies |
|
Lysosomal disorders |
Anemia, pancytopenia,abnormal liver function, CK,urine GAG
elevation, Cherryred spot / pigmentary changesin retina,
dysostosis multiplex |
Complete blood count, liver function, renal function, serumcreatine
kinase, Ophthalmicfundus exam, skeletal survey
|
Urine MPS and oligosaccharide screen, specific
enzyme assay, plasma chitotriosidase, gene studies
|
|
Peroxisomal disorders |
Deranged liver enzymes andVLCFA, abnormal lipids |
Liver function tests, Renalfunctions, Lipid profile, |
MRI brain, VLCFA analysis, Phytanic and pristanic
acids, Plasmalogenes, gene studies Complementation studies,
|
|
Disorders of neuro-transmitter metabolism |
Abnormal amino acid, organic acid profile, lipid profile, hyperprolactinemia,
increased pipecolic acid |
GELAK, S. Prolactin, S. uricacid, homocysteine, S. folic acid,vit
B12 levels
|
Paired sampling – plasma and CSF amino acids, glucose
andlactate, CSF neurotransmittersand pterins, gene studies |
|
Disorders in the meta-bolism of vitamins and (non-protein)
cofactors |
Abnormal GELAAK, S. Pro-lactin,S. Uric Acid, hyperhomo-cysteinemia,
with normal S. folic acid, vit B12 levels |
Testing for GELAK, S. Prolactin,S. Uric Acid, homocysteine, S.
folicacid, vit B12 levels
|
TMS, urine GC-MS, Biotinidase enzyme, gene studies |
|
Disorders in the meta-bolism of trace elements and metals |
Abnormal liver enzymes, KayserFletcher -ring,
S.Ceruloplasmin,Serum and urinary copper (Wilson, Menke),
S.Ferritin, S.uric acid (MoCD) |
LFT, Eye exam for KayserFletcher-ring, testing
forS.Ceruloplasmin, S.Ferritin,S.uric acid (MoCD)
|
Serum and urinary copper (Wilson, Menke), Hair shaft
examination pili torti (Menke), gene studies
|
|
Disorders and variants in the metabolism of xeno-biotics |
Trimethylaminuria, White matterand other parenchymal abnor-malities
in brain (Sjogren Larson) |
Specific punjent odor in urine(trimethylaminuria)
|
MRI brain,Skin fibroblast enzyme assay (SLS), freeTMA in urine,
gene studies |
|
SSIEM: Society for study of inborn errors of metabolism;
mtDNA: mitochondrial DNA; nDNA: nuclear DNA; FAB-MS: Fast atom
bombardment – mass spectrometry; MoCD: Molybdenum Cofactor
Deficiency; TMA: trimethylaminuria; * Common only. |
Enzyme Assay
Testing for activity of an enzyme requires
recreating/simulating the actual enzymatic reaction outside of body
using appropriate body fluids/tissue (leukocytes, plasma/serum, skin
fibroblasts or liver biopsy specimens), and using artificial or natural
substrates [15].
Lysosomal storage disorders: More than 24 enzymes
are known, and can be tested in blood (leucocytes, sometimes plasma) and
cultured skin fibroblasts. Examples are enzymes for Gaucher disease,
Niemann Pick disease [13]. Usual requirement for testing is 4-5 mL blood
in heparin vial (or sometimes EDTA vial). Lysosomal enzymes can also be
measured on a dried blood spot [14]
Galactosemia: Galactose-6-phosphate uridyl
transferase (GALT), Galactokinase (GALK), and Galactose-1-phosphate
uridyl epimerase (GALE) can be performed on red blood cells (RBCs) only;
therefore, blood in heparin vacutainers is required. Semi-quantitative
testing can also be performed on dried blood spots (Beutler spot test),
and is useful in newborn screening.
Biotinidase deficiency: This enzyme assay can be
performed in serum (quantitative) or on dried blood spots (semi
quantitative), and is useful in newborn screening.
Other small molecule disorders: Enzyme assays for
fatty acid oxidation defects (FAOD), gluconeogenesis (hereditary
fructose intolerance, fructose 1-6-biphosphatase ), glycogen storage
disorders (GSD), urea cycle defects or organic acidurias, tyrosinemia,
cystinosis, and porphyrias used to be the standard diagnostic modalities
until molecular diagnosis became widely available. There are limited
accredited laboratories for enzyme assays [16]. Majority of these
disorders would require specialized tissues such as cultured skin
fibroblasts, frozen liver biopsy or muscle biopsy specimen [6]. The
diagnosis of these disorders relies heavily upon specific metabolite
analysis using methods described below and molecular genetic testing
involving sequencing of particular gene or group of genes.
Detection of Metabolites
These tests detect specific metabolites through
simple chemical reactions with precision, sometimes alleviating the need
for molecular genetic testing. Urine metabolic tests can be done easily
in most laboratories. Table II depicts the
preliminary simple bedside tests that can be used for the diagnosis of
inborn metabolic errors. Among the specialized biochemical tests, three
methods are described in more detail; whereas, other tests are mentioned
in Table III.
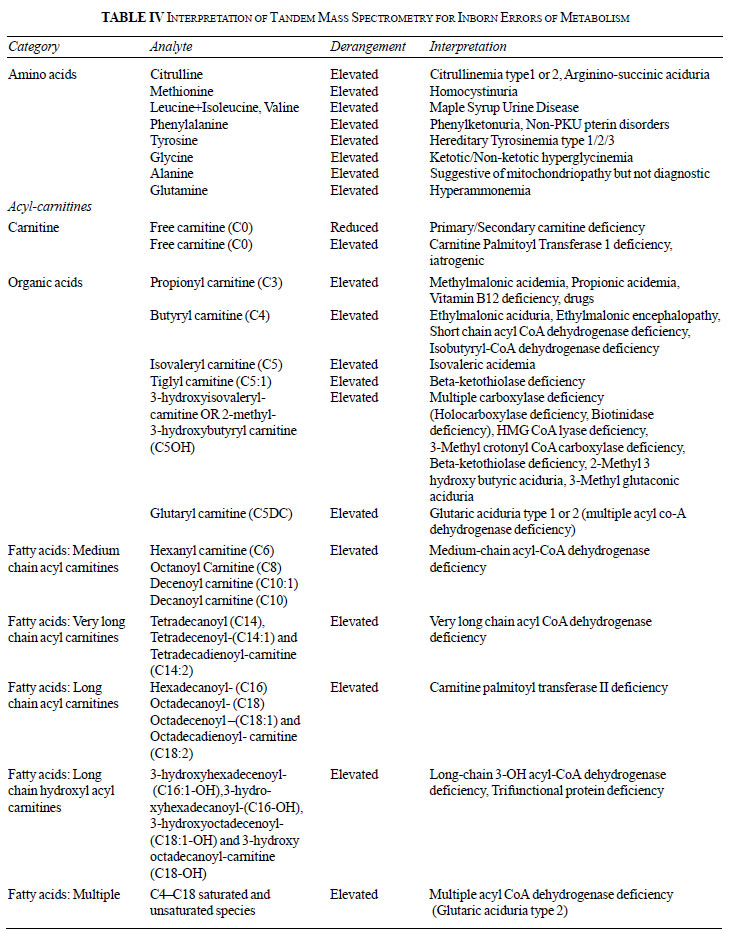
Tandem mass spectrometry
Tandem mass spectrometry (TMS), also known as mass
spectrometry-mass spectrometry (MS/MS) as two mass spectrometers are
situated in tandem. This is a revolutionary technology devised in early
90s [17], which can detect multiple independent metabolites/analytes in
a single test. The basic principle of TMS relies on the ionization and
fragmentation of each molecule or metabolite into specific ions coupled
with a robust detection system which is computerized to provide results
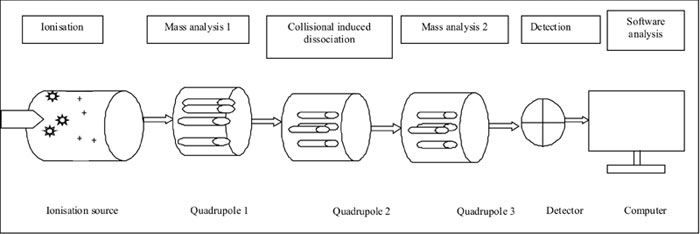
[18]. Fig. I provides a simplified diagrammatic
representation of MS/MS equipment [19]. A sample is first ionized and
made to pass through a series of chambers, which serve specific purpose
of further ionization, collision-induced dissociation and further
ionization of daughter ions before reaching a detection chamber that
recognizes individual ions based on specific mass-to-charge ratio. The
computer then interprets the data and provides highly accurate results
of each analyte. The utility of a TMS is multi-fold and its applications
are expanding. It measures amino-acids, organic acids and fatty acids in
form of their acyl-carnitine esters. This particular technology forms
the core of expanded newborn screening throughout the developed world
[20]. Table IV provides interpretations of few
metabolites [21]. TMS does not screen for many IEMs such as
mitochondriopathies, purine and pyrimidine disorders, neurotransmitters,
congenital disorders of glycosylation (CDG) and very long chain fatty
acids. TMS using dried blood spot is highly sensitive but not specific,
and thus remains a screening test for majority of disorders. Its use for
prenatal diagnosis is not recommended. Higher efficacy equipments such
as liquid chromatography-MS/MS (LC-MS/MS) are now employed for better
accuracy in biological samples, including CSF and plasma.
 |
| |
 |
|
Fig. 1 Basic steps of tandem mass
spectrometry.
|
GC-MS urinalysis for organic acids
GC-MS is gas chromatography-mass spectrometry
technology to detect metabolites specific for small molecule disorders
(typically organic acids and related) in the urine [22]. The organic
acids are volatile and tend to evaporate without preservation, which
requires samples to be frozen. Detection of organic acids in urine
should ideally be done in frozen sample or reliably if transported under
ambient temperatures not exceeding 25°C. Transportation is stable when
urine sample is put on a special grade filter paper that is able to soak
in all metabolites and fix or stabilize it preventing it from
evaporation [22]. They can be eluted back from filter paper into liquid
solution using simple techniques. Interpretation of results of urine
organic acid analysis by GC-MS requires expertise and biochemical
background training, and thus should be taken up only by specialized
genetic laboratories or institutes. The modality can also measure
metabolites in bile, plasma and other body fluids including postmortem
samples when there is a failure to collect urine.
Ultra/high performance liquid chromatography (U/HPLC)
or ion-exchange chromatography
The measurement of specific amino acids in body
fluids such as plasma/serum, urine or CSF is being utilized widely for
diagnosing many amino-acid disorders such as phenylketonuria,
tyrosinemia type 1/2/3, maple syrup urine disease, and homocystinuria (methionine
as well as homocysteine which requires a different algorithm for
identification) [23]. There are multiple methods for quantifying amino
acids – chromatographic as well as electrophoresis. Most laboratories
currently employ either HPLC or UPLC technique because of the relative
ease and high specificity. All samples (plasma not whole blood, urine or
CSF) for chromatography or electrophoresis should be transported in
frozen conditions using dry ice. Samples exposed to high temperatures
and harsh conditions would destroy the amino acids and give erroneous or
inaccurate results. Samples can also be collected in a sulfosalicylic
acid (SSA) pretreated vial to ensure stability. CSF samples should be
paired with plasma samples for accurate interpretation. Diagnosis of
non-ketotic hyperglycinemia will be made when both CSF and plasma levels
of glycine are elevated. Isolated elevation of glycine may be due to
other non-genetic causes such as perinatal asphyxia or bloody tap (due
to contamination of CSF with blood). Interpretation for some common and
important metabolite/amino acid derangement is provided in Table
III. Detection of amino acids qualitatively in the urine (and
sometimes both urine as well as plasma) has also been used traditionally
for the diagnosis amino acid disorders. The test is done by a thin-layer
chromatography technique that is a simple test using filter paper. The
pattern of excretion of amino acids is interpreted to get to a
diagnosis.
Other Metabolic Investigations
Neurotransmitter assays: This is a specialized
test performed only on CSF using sophisticated LC-MS/MS and GCMS
technique [10] that only few laboratories have standardized and thus
authorized to perform.
Serum transferrin isoforms for glycosylation pattern:
This is a widely used screening test for CDGs, the diagnosis of which
requires genetic testing. The test was initially established using iso-electric
focusing (IEF) [24] followed by capillary zone electrophoresis (CZE) in
the nineties [25]. There are two abnormal patterns of transferrins
identified in CDGs leading to their classification into either CDG type
1 (showing a type 1 pattern of derangement of transferrin isoforms) or
type 2. However, not all CDGs would show up with these patterns [26].
The test can also be done using HPLC, MALDI-TOF-MS/MS or LC-MS/MS [27].
Metabolic biomarkers: Testing for chitotriosidase,
CCL18/PARC, heparin thrombin cofactor ii, lyso GB3 are available in few
specialized laboratories. Multiple biomarkers are in use for diagnosis
and prognosis of many metabolic disorders. A holistic approach is
recommended to detect and interpret certain (specific) pattern of
metabolites in body fluids to make a metabolic diagnosis. This new
system also known as the metabolomics approach is rapidly advancing and
holds promise for the future [28]
Molecular Genetic Testing
The molecular genetic test entails testing of either
single or multiple genes together depending upon the suspicion of the
disease. For disorders where a specific diagnosis is already made using
other techniques such as GALT deficiency, only one gene (GALT)
may be tested. However, for other disorders like mitochondrial
disorders, multiple genes are to be tested together for lack of evidence
towards any specific genes (barring few exceptions). There are two
methods employed for molecular genetic testing: a traditional, well
established and more precise method of Sanger sequencing [29], and a
more advanced method known as Next generation sequencing (NGS). Both
methods allow for sequencing of genes, determining the sequence of base
pairs in the DNA of the exons/exon-intron boundaries or coding regions
of a gene. However, Sanger sequencing is time consuming, laborious and
expensive as it is performed fragment wise, one by one for each fragment
of a gene. The NGS utilizes the multiplexing of all the sequencing
fragments and thus has the capacity to sequence any number of DNA
fragments simultaneously. The NGS can sequence from one gene to whole
exome or even genome which is about 3 billion base pairs. By
multiplexing the sequencing there is considerable saving of time as well
as cost. It is now becoming the standard diagnostic methodology in most
of genetic laboratories, including for metabolic disorders.
Pitfalls
An entire battery of test has been developed over the
years to diagnose IEMs. The basic and advanced metabolic test would
diagnose majority of IEMs, except few. TMS can detect only three groups
of disorders (fatty acid oxidation, organic and few amino acidurias) and
not mitochondrial disorders. A biochemical test may show normal
metabolite levels as they are not always deranged. In such cases, the
metabolic tests should be repeated during acute sickness.
Conclusion
Inborn errors of metabolism are disorders often
encountered in pediatric emergencies presenting with common symptoms. As
many of them are treatable, a high index of suspicion can lead to their
early recognition. Early and accurate treatment is the key to a fruitful
outcome. IEMs can be screened by basic biochemical tests. Appropriate
samples should also be collected and stored at the time of acute illness
for advanced genetic tests, which can be carried out later. All IEMs
have a genetic basis: hence, an accurate genetic counseling should be
provided to the families for prevention in future births. With knowledge
of the basic and advanced metabolic tests, the pediatrician would be
empowered to diagnose and treat most IEMs.
|
Key Messages
• Inborn errors of metabolism (IEM) may
present at any age (antenatal, birth, infancy, childhood or
adulthood), with common illnesses such as vomiting, respiratory
distress, lethargy or even seizures.
• A recurrence of similar illness in the past
or in family should raise suspicion for an IEM. Presence of
consanguinity must be asked in all cases.
• In an acutely sick child, IEMs should be
suspected at the same time as other diagnoses like infection,
trauma, poisoning. Prompt testing with basic metabolic tests
would save time and benefit the child with early treatment
possibilities.
• Communication with metabolic (biochemical
and genetic) laboratory and metabolic specialist is beneficial
for appropriating the testing methodology as well as in acute
management.
|
References
1. Sample Registration System (SRS) Bulletin. Office
of Registrar General of India (for the year 2017). 2019; 52 (1).
Available From: censusindia.gov.in/vital_statistics/SRS_Bulletins/SRS_Bulletin-Rate-2017-_May_2019.pdf.
Accessed May 24, 2019.
2. Garrod AE. Inborn errors of metabolism. In:
Frowde H, Hodder and Stoughton, editors. The Croanian Lectures Delivered
Before the Royal College of Physicians of London in June 1908. London:
Oxford University Press; 1909.
3. Applegarth DA, Toone JR, Lowry RB. Incidence of
inborn errors of metabolism in British Columbia, 1969-1996. Pediatr.
2000;105:e10.
4. Bijarnia-Mahay S, Arora V, Puri RD, Lall M, Saxena
R, Verma J, et al. The changing scenario in prenatal diagnosis of
genetic disorders: Genetics to genomics. Curr Med Res Prac. 2018; 8:
203-8.
5. Vernon HJ. Inborn errors of metabolism: Advances
in diagnosis and therapy. JAMA Pediatr. 2015;169:778-82.
6. Scriver CR, Beaudet AL, SlyWS, Valle D, Vogelstein
B, editors. The Online Metabolic and Molecular Bases of Inherited
Disease (OMMBID). New York: McGraw-Hill; 2018.
7. Blau N, Duran M, Gibson KM, Dionisi-Vici C,
editors. Physician’s Guide to the Diagnosis, Treatment, and Follow-up of
Inherited Metabolic Diseases.Berlin: Springer Nature; 2014.
8. Saudubray JM, van den Berghe G, Walter J, editors,
6th ed. Inborn Metabolic Diseases: Diagnosis and Treatment. Berlin:
Springer; 2016.
9. Nyhan WL. When to suspect metabolic disease. In:
Hoffmann GF, Zschocke J, Nyhan WL Eds. Inherited Metabolic Diseases: A
Clinical Approach. 2nd ed. Berlin: Springer; 2017. p.19-28.
10. El-Hattab AW. Inborn errors of metabolism. Clin
Perinatol. 2015; 42:413-39.
11. Gibson K, Duran M. Simple metabolic screening
tests. In: Blau N, Duran M, Gibson K, editors. Laboratory Guide
to the Methods in Biochemical Genetics. Berlin: Springer; 2008. p.
22-33.
12. Garg U, Smith LD, Heese BA. Introduction to the
laboratory diagnosis of inherited metabolic diseases. In: Garg U,
Smith LD, Heese BA, editors. Laboratory Diagnosis of Inherited Metabolic
Diseases. Washington: AACC press; 2012. p. 1-12.
13. Clayton PT. Inborn errors presenting with liver
dysfunction. Semin Neonatol. 2002; 7:49-63.
14. Alam S, Lal BB. Metabolic liver diseases
presenting as acute liver failure in children. Indian Pediatr.
2016;53:695-701.
15. Verma J, Thomas DC, Kasper DC, Sharma S, Puri RD,
Bijarnia-Mahay S, et al. Inherited metabolic disorders: Efficacy
of enzyme assays on dried blood spots for the diagnosis of lysosomal
storage disorders. JIMD Rep. 2017;31:15-27.
16. Bijarnia-Mahay S, Movva S, Gupta N, Sharma D,
Puri RD, Kotecha U, et al. Molecular diagnosis of hereditary
fructose intolerance: Founder mutation in a community from India. JIMD
Rep. 2015;19:85-93.
17. Millington DS, Kodo N, Norwood DL, Roe CR. Tandem
mass spectrometry: A new method for acylcarnitine profiling with
potential for neonatal screening for inborn errors of metabolism. J
Inherit Metab Dis 1990;13:321-4.
18. Ho CS, Lam CW, Chan MH, Cheung RC, Law LK, Lit
LC, et al. Electrospray ionization mass spectrometry: Principles
and clinical applications. Clin Biochem Rev. 2003; 24:3-12.
19. Mass Spectrometry- Atomic Particles Stored in a
Cage Without Material Walls. Avialable from:URL:https://www.biologie.hu-berlin.de/de/gruppenseiten/oekologie/meth/massspec/
mass.sp. Accessed May 24, 2019.
20. Wilcken B. Recent advances in newborn screening.
J Inherit Metab Dis. 2007;30:129-133.
21. Lehotay DC, Hall P, Lepage J, Eichhorst JC, Etter
ML, Greenberg CR. LC-MS/MS progress in newborn screening. Clin Biochem.
2011;44:21-31.
22. Fu X, Iga M, Kimura M, Yamaguchi S. Simplified
screening for organic acidemia using GC/MS and dried urine filter paper:
a study on neonatal mass screening. Early Hum Dev. 2000;58:41-55.
23. Narayan SB, Ditewig-Meyers G, Graham KS, Scott R,
Bennett MJ. Measurement of plasma amino acids by Ultraperformance®
Liquid Chromatography. Clin Chem Lab Med. 2011; 49:1177-85.
24. Jaeken J, van Eijk HG, van der Heul C, Corbeel L,
Eeckels R, Eggermont E. Sialic acid-deficient serum and cerebrospinal
fluid transferrin in a newly recognized genetic syndrome. Clin Chim Acta 1984;
144:245-7.
25. Prasad R, Stout RL, Coffin D, Smith J. Analyses
of carbohydrate deficient transferrin by capillary zone electrophoresis.
Electrophoresis. 1997;18:1814-8.
26. Jaeken J. Congenital disorders of glycosylation.
Ann N Y Acad Sci. 2010;1214:190-8.
27. Li X, Raihan MA, Reynoso FJ, He M. Glycosylation
analysis for congenital disorders of glycosylation. Curr Protoc Hum
Genet. 2015;86:1-22.
28. Tebani A, Abily-Donval L, Afonso C, Marret S,
Bekri S. Clinical metabolomics: The new metabolic window for inborn
errors of metabolism investigations in the post-genomic era. Int J Mol
Sci. 2016;17.pii:E1167.
29. Sanger F, Nicklen S, Coulson AR. DNA sequencing
with chain-terminating inhibitors. Proc Natl Acad Sci USA.
1997;74:5463-7.
|
|
|
 |
|