|
|
|
Indian Pediatr 2017;54:
735-738 |
 |
Pediatric Rhabdomyosarcoma in India: A
Single-center Experience
|
|
Deepak Bansal, Anirban Das, Amita Trehan, *Rakesh
Kapoor, #Naresh K
Panda, $Radhika
Srinivasan, ‡Nandita
Kakkar, ^Kushaljit
S Sodhi, ^Akshay
K Saxena and **Katragadda Lakshmi Narasimha Rao
From Pediatric Hematology/Oncology unit, Advanced
Pediatrics Center, *Department of Radiotherapy, #Otolaryngology, $Cytology,
‡Histopathology, ^Radiodiagnosis, and **Pediatric
Surgery, Postgraduate Institute of Medical Education and Research,
Chandigarh, India.
Correspondence to: Dr Deepak Bansal. Professor,
Hematology-Oncology unit, Department of Pediatrics, Advanced Pediatrics
Center, Postgraduate Institute of Medical Education and Research,
Chandigarh, India.
Email:
[email protected]
Received: October 01, 2016;
Initial review: December 27, 2016;
Accepted: July 07, 2017.
|
Objective: Analyze the profile and outcome of children with
rhabdomyosarcoma from a pediatric-oncology unit.
Design: Retrospective analysis
of case records over 23 years (1990-2012).
Setting: Government-run,
tertiary-care, university hospital in Northern India.
Participants: 159 children
(<12-years) with a diagnosis of rhabdomyosarcoma were enrolled. The
median age was 4 years; 13% were infants.
Main outcome measure: Five-year
event free survival.
Results: The median symptom
interval was 2-months. Head and neck region was the most frequent site
(44%), followed by tumors in the extremity (15.7%). The majority (67%)
of the tumors were located at ‘unfavorable’ sites; 68% were >5 cm in
size. The most frequent (58%) pathological subtype was embryonal.
Treatment was based on the ‘Intergroup Rhabdomyosarcoma Study (IRS)
Group’ risk-stratification. 33% were ‘low-risk’ children, 11% were
‘high-risk’. Treatment-refusal (18%) and abandonment (33%) were major
impediments. The median ± SE five-year event free survival of those
taking treatment was 43.6 ± 6%.
Conclusions: Large sized tumors,
tumors at unfavorable locations, and treatment refusal/abandonment
contributed to inferior outcome in children with rhabdomyosarcoma.
Keywords: Outcome, Sarcoma, Survival,
Treatment refusal.
|
|
R
habdomyosarcoma (RMS) is the most common
soft-tissue sarcoma in children, with an annual incidence of 4.3/million
below the age of 20 years [1]. Multimodality treatment has resulted in a
5-year survival of approximately 67% [2]. The current focus is on
reduction of treatment-related toxicity, improving cure-rates for
relapsed disease by inclusion of novel agents, and identification of
newer prognostic factors for risk-adapted therapy [3].
There is limited data on the outcome of this disease
from developing countries, including India [4-6]. The multidisciplinary
approach mandated for managing RMS is often a hindrance for orchestrated
treatment, as well as data collection in pediatric oncology units in
developing countries. Selected studies from India have reported
sub-optimal survival (13-25%) [4,5]. The factors contributing to poor
survival include, treatment abandonment, infection related mortality,
lack of surgical expertise for local disease-control, and failure to
deliver chemotherapy in a timely manner due to poor compliance [4-6].
The aim was to analyze the profile and outcome of children with RMS from
a pediatric oncology unit. The purpose was to characterize the disease
profile in our population and to identify hindrances for improving
survival.
Methods
The study was performed in a government-run,
tertiary-care, university hospital in Northern India. A retrospective
analysis of case-records of children (age <12 years at diagnosis) with
RMS presenting to the pediatric-oncology clinic over a 23-year
(1990-2012) period was performed after Institutional Ethical Clearance.
The patients were classified by the ‘Intergroup Rhabdomyosarcoma Study
(IRS) Group’ staging and grouping system as low, intermediate or
high-risk [7]. The IRS ‘group’ was determined after the initial surgical
procedure, prior to systemic therapy and was based on the extent of
residual tumor after surgery with consideration of regional lymph node
involvement. The IRS ‘stage’ was based on tumor size, invasiveness,
nodal status, and the site of primary tumor. The group and stage were
both taken into consideration for the final risk stratification. The
favorable sites included orbit, superficial head and neck, biliary tree,
vagina, and para-testis; the remaining sites are considered unfavorable
[7,8]. The tumor size was based on the largest dimension of the primary
tumor reported in the pretreatment imaging. Nodal involvement was based
on physical examination, imaging studies and/or node sampling at
surgery. Pathology was classified as ‘embryonal,’ ‘alveolar,’ ‘pleomorphic’
or ‘not otherwise specified (NOS).’ Molecular tests were not available.
Staging investigations included a computed tomography (CT) of the chest
and a bone scan. Cerebrospinal fluid (CSF) was examined for
para-meningeal tumors. A bone marrow examination was performed for
noticeable hematological abnormality, or in the presence of other sites
of metastasis. Congenital RMS was defined as presentation within the
first month of life with history of onset of symptoms since birth [9].
Treatment included neo-adjuvant chemotherapy,
followed by surgical excision (wherever feasible), radiotherapy (for
residual disease, if any, and, for inoperable tumors), and adjuvant
chemotherapy, based on the Children’s Oncology Group (COG) protocols
[7]. Low-risk patients received vincristine and actinomycin-D for 22
weeks, while intermediate and high-risk groups were treated with
vincristine, actinomycin-D, and cyclophos-phamide for the duration of
43-weeks. The specific high-risk protocol was not administered due to
apprehension regarding toxicity. The response was evaluated by
radiological imaging (CT/Magnetic Resonance Imaging) following week 9-12
of chemotherapy. Survival estimates were calculated using the
Kaplan-Meier method. Analysis was performed with SPSS v20.0 (IBM).
Results
The data of 159 patients was collected. The median
age at presentation was 4 years (Interquartile range, IQR 2;7). There
were 20 (13%) infants; 5 (3%) had congenital rhabdomyosarcoma. Nine (6%)
children were >10 years at presentation. The male:female ratio was
2.7:1. The median symptom interval was 2 months (IQR 1;5). Head and neck
region was the most frequent site (44%), followed by tumors in the
extremity (15.7%) (Table I). Majority (67%) of the tumors
were located at unfavorable sites. Size was documented in 101 patients;
69 (68%) were >5 cm. Nodal status was documented in 76 and was positive
in 36 (47%). Thirteen (8%) had evidence of metastasis at diagnosis. The
most frequent (58%) pathological subtype was embryonal. Information
required for risk-stratification was available for 126; low-risk: 42
(33%), intermediate-risk: 70 (56%) and high-risk: 14 (11%) [7]. Of the
129 patients in whom treatment was initiated, surgery was performed in
63 (49%), while 54 (42%) received radiotherapy; 31 (24%) received both.
TABLE I Details of Patients (N=159) with Rhabdomyosarcoma
|
Characteristic |
No. (%) |
|
Site |
|
Para-meningeal |
33 (20.8) |
|
Extremity |
25 (15.7) |
|
Orbit |
20 (12.6) |
|
Head and neck: Others |
17 (10.8) |
|
Bladder |
16 (10) |
|
Pelvic/Abdomino-pelvic |
15 (9.4) |
|
Genitourinary(non-bladder/prostate) |
13 (8.2) |
|
Trunk |
11 (6.9) |
|
Retroperitoneal |
3 (1.8) |
|
Biliary |
2 (1.3) |
|
Others |
5 (3.1) |
|
Histological subtypes |
|
Embryonal |
92 (57.9) |
|
Botryoid variant |
6 (3.8) |
|
Alveolar |
8 (5) |
|
Pleomorphic |
6 (3.8) |
|
Not otherwise specified |
47 (29.6) |
Caregivers of 28 (18%) patients refused treatment at
the outset. In addition, 52 (33%) abandoned treatment at various phases
of therapy. The remaining had a mean (SD) follow-up duration of 4.4
(2.6) years (range: 0.5-10), including two patients with ongoing
treatment.
Among the 77 patients who completed treatment, there
were 36 survivors, 7 with progressive disease, 5 deaths due to febrile
neutropenia, and 29 relapses at a median duration of 11 months (IQR
7;15) following diagnosis. The latter received palliative care. Median ±
SE five-year event-free survival (EFS) after excluding patients with
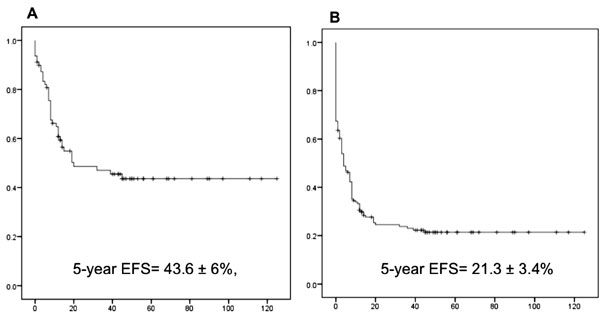
treatment refusal/abandonment was 43.6 ± 6%. When abandonment/refusal
were included as events, the 5-year EFS dropped to 21.3 ± 3.4% (Fig.
1). The last two years (2011-12) demonstrated a similar rate of
refusal/abandonment (54% for 2011-12 versus 50% for 1990-2010).
The survival was site-dependent; favorable sites, including orbit (EFS
57.1 ± 1.8%) demonstrated superior survival as compared to the following
primary sites: parameningeal (35.2 ± 1.5%) extremities (36 ± 1.6%) and
bladder-prostate (22.2 ± 1.2 %) (P=0.03).
 |
|
Fig. 1 Five-year event-free
survival in childhood rhabdomyosarcoma: (a) Excluding treatment
refusal/abandonment; (b) Including treatment refusal/abandonment
as events.
|
Discussion
The study reports 159 children with RMS over a
23-year (1990-2012) period. Several tumors were >5 cm in size at
presentation. The majority were located at unfavorable sites. Treatment
refusal and abandonment were major concerns. Overall, a 5-year EFS of
43.6 ± 6% was documented. Orbit as the primary site demonstrated
superior survival as compared to parameningeal, extremities and
bladder-prostrate primaries.
The limitations of the study include the extended
duration of enrollment, precluding uniformity in diagnostic and
therapeutic modalities. In addition, high-risk protocol was not
administered for likelihood of increasing treatment related mortality.
Treatment refusal and abandonment were major predicaments.
The clinical and investigational profile of our
patients was comparable to that reported from the West. Prevalence in
infants (13%) was similar to previous reports (5-10%) [9]. The median
age was consistent with prior SEER (Surveillance, Epidemiology and
End-results Program) data, as well as previous reports from India [4-6,
10]. A size exceeding 5 cm
has been demonstrated to be predictive of mortality [11].
This could plausibly imply a delayed referral.
However, the impact of a delayed diagnosis on the outcome in RMS is
ambiguous, plausibly due to the intrinsic biology of the tumor [12]. It
is hypothesized that early presentation might signify a more aggressive
biology. The proportion of tumors at unfavorable sites (67%) in this
study was akin to that reported in a multi-institutional study (55%)
[13]. The pathology was embryonal in 58% [14]. Pleomorphic RMS, rarely
reported in children (1%), was described in 4% [13].
RMS-NOS was reported in 30%; previous reports
noted a lower frequency (13%). This could plausibly be attributed to the
pathologists’ inability at accurate characterization [13].
Earlier studies from India have reported inferior
remission (25%) and cure-rates (13%) [4,5]. Survival in other developing
countries, including Nigeria were sub-optimal (28%) [15].
More recent studies have reported encouraging
results. Salman, et al. [16] from Lebanon reported a 5-year
disease-free survival of 64%.
A multi-centric study from China reported 10-year EFS of
53.4 ± 5.1% [17]. In 2012, Dua, et al. [6] from India reported an
improved 5-year EFS of 57.1 ± 13.2% in a limited numbers of patients.
The survival is superior in the more developed Asian countries,
including Korea (5-year EFS: 59%) and Singapore (5-year EFS: 75%)
[18,19]. Non-adherence to treatment has been previously reported from
the two studies from India (30% and 60%) [4,5], and has been a major
cause of inferior outcome in the developing world [20,21].
A complex interplay of biological, socio-economic
and treatment-related factors underlie the challenge [21].
In conclusion, in comparison to the West, children
with RMS from India had an inferior survival despite similar disease
characteristics. Treatment refusal/abandonment were recognized as major
impediments to improving outcome. The measures incorporated in the unit
in the recent years to reduce abandonment include, repetitive counseling
sessions with the extended family as stakeholders, active collection of
government funds with the help of social workers provided by
non-government organizations, support for nutrition, as well as tracking
of defaulters. Incorporation of measures to improve survival need to be
studied by future researchers.
Funding: None. Competing interests: None
stated.
|
What is Already Known?
•
Five-year survival for pediatric
rhabdomyosarcoma is sub-optimal in Indian centers.
What This Study Adds?
• 5-year Event-free
Survival of 43.6 ± 6% was observed in Indian patients who
received complete treatment.
•
Large sized tumors (68%), tumors at unfavorable location
(67%), treatment-refusal (18%) and abandonment (33%) were
identified as reasons for the sub-optimal outcome.
|
References
1. Wexler LH, Meyer WH, Helman LJ. Rhabdomyosarcoma.
In: Pizzo PA, Poplack DG. Principles and practice of pediatric
oncology. 6th ed. Philadelphia: Lippincott Williams and Wilkins; 2011.
p.923.
2. Gatta G, Botta L, Rossi S, Aareleid T,
Bielska-Lasota M, Clavel J, et al; EUROCARE Working Group.
Childhood cancer survival in Europe 1999-2007: results of EUROCARE-5 – a
population-based study. Lancet Oncol. 2014;15:35-47.
3. Raney RB, Anderson JR, Barr FG, Donaldson SS,
Pappo AS, Qualman SJ, et al. Rhabdomyosarcoma and
undifferen-tiated sarcoma in the first two decades of life: A selective
review of intergroup rhabdomyosarcoma study group experience and
rationale for intergroup rhabdomyosarcoma study V. J Pediatr Hematol
Oncol. 2001;23:215-20.
4. Thavaraj V, Kumar R, Dawar R, Gupta DK, Mohanti
BK, Singh R, et al. Treatment outcome and survival of
rhabdomyosarcoma (RMS) in children. Med Pediatr Oncol. 2001;37:286.
5. Mehta P, Swami A, Chaphekar M, Currimbhoy Z,
Agarwal B. Soft tissue sarcoma (STS): Perspective from a paediatric
cancer unit (PCU) at a trust hospital in Mumbai, India. Pediatr Blood
Cancer. 2004;43:475.
6. Dua V, Yadav SP, Prakash A, Sachdeva A.
Encouraging treatment outcomes of pediatric rhabdomyo-sarcoma: a developing
world experience. Pediatr Hematol Oncol. 2012;29:677-8.
7. Malempati S, Hawkins DS. Rhabdomyosarcoma: review
of the Children’s Oncology Group (COG) Soft-Tissue Sarcoma Committee
experience and rationale for current COG studies. Pediatr Blood Cancer.
2012;59:5-10.
8. Womer RB, Barr FG, Linardic CM. Rhabdomyosarcoma.
In: Orkin SH, Fisher DE, Ginsburg D, Thomas Look A, Lux SE,
Nathan DG. Nathan and Oski’s Hematology and Oncology of Infancy and
Childhood. 8th ed. Philadelphia: Elsevier Saunders; 2015. p.1929.
9. Ferrari A, Casanova M, Bisogno G, Zanetti I,
Cecchetto G, De Bernardi B, et al. Rhabdomyosarcoma in infants
younger than one year old: a report from the Italian Cooperative Group.
Cancer. 2003;97:2597-604.
10. Ognjanovic S, Linabery AM, Charbonneau B, Ross
JA. Trends in childhood rhabdomyosarcoma incidence and survival in the
United States, 1975-2005. Cancer. 2009;115:4218-26.
11. Ferrari A, Miceli R, Casanova M, Meazza C, Favini
F, Luksch R, et al. The symptom interval in children and
adolescents with soft tissue sarcomas. Cancer. 2010;116:177-83.
12. Ferrari A, Lo Vullo S, Giardiello D, Veneroni L,
Magni C, Clerici CA, et al. The Sooner the better? How symptom
interval correlates with outcome in children and adolescents with solid
tumors: Regression Tree analysis of the findings of a prospective study.
Pediatr Blood Cancer. 2016;63: 479-85.
13. Sultan I, Qaddoumi I, Yaser S, Rodriguez-Galindo
C, Ferrari A. Comparing adult and pediatric rhabdomyosarcoma in the
surveillance, epidemiology and end results program, 1973 to 2005: an
analysis of 2,600 patients. J Clin Oncol. 2009;27:3391-7.
14. Maurer HM, Gehan EA, Beltangady M, Veneroni L,
Magni C, Clerici CA, et al. The Intergroup Rhabdomyosarcoma
Study-II. Cancer. 1993;71:1904-22
15. Uba FA, Chirdan LB. Clinical characteristics and
outcome of surgical treatment of childhood rhabdomyosarcoma: A 7-year
experience. Afr J Paediatr Surg. 2008;5:19-23.
16. Salman M, Tamim H, Medlej F, El-Ariss T, Saad F,
Boulos F, et al. Rhabdomyosarcoma treatment and outcome at a
multidisciplinary pediatric cancer center in Lebanon. Pediatr Hematol
Oncol. 2012;29:322-34.
17. Ma X, Huang D, Zhao W, Sun L, Xiong H, Zhang Y,
et al. Clinical characteristics and prognosis of childhood
rhabdomyosarcoma: A ten-year retrospective multicenter study. Int J Clin
Exp Med. 2015;8:17196-205.
18. Park JA, Kim EK, Kang HJ, Shin HY, Kim IH, Ahn
HS. Initial response to treatment was highly associated with the
prognosis of childhood rhabdomyosarcoma: a retrospective analysis of a
single center experience in Korea. Cancer Res Treat. 2008;40:111-5.
19. Aung L, Soe TA, Chang KT, Quah TC. Singapore
rhabdomyosarcoma (RMS) experience: Shall we change our practice? Ann
Acad Med Singapore. 2014;43:86-95.
20. Arora RS, Pizer B, Eden T. Understanding refusal
and abandonment in the treatment of childhood cancer. Indian Pediatr.
2010;47:1005-10.
21. Arora RS, Eden T, Pizer B. The problem of
treatment abandonment in children from developing countries with cancer.
Pediatr Blood Cancer. 2007;49:941-6.
|
|
|
 |
|
