|
Shwachman-Diamond Syndrome
(SDS) is a rare inherited disorder characterized by pancreatic
insufficiency, bone marrow dysfunction and skeletal abnormalities
[1]. It is the second most common cause of pancreatic insufficiency
in children, next to cystic fibrosis, and probably the third most
common inherited bone marrow failure syndrome after Fanconi’s anemia
and Diamond-Blackfan anemia [2]. The estimated incidence in the West
is approximately 1: 100,000 to 1: 200,000 live births [3] and till
date there are only two case reports of SDS from India [4,5].
Considering the heterogeneity of the disease associated phenotypic
manifestations, and lack of awareness, it is likely that we are
missing many cases in India. We report a case of classical SDS who
is doing reasonably well on pancreatic enzyme supplement for two
years.
Case report
A 11-year-7 month-old boy presented to us with
chronic diarrhea (greasy stools, 5 to 10 times a day) and failure to
thrive since birth. He also had frequent episodes of fever requiring
antibiotics, recurrent oral ulcers, pruritic papular skin rash and
dental caries. The child was a product of non-consanguineous
marriage with normal birth weight and an apparently healthy elder
sister. There was no family history of pancreatic insufficiency or
hematological disorders.
His physical examination revealed growth
retardation (weight 17.5 kg, height 117 cm; both <5 th
percentile for age), dental caries, hyperpigmented scar marks of
skin lesions, and a just palpable soft liver (span 8cm) without
splenomegaly. Investigations done elsewhere 2 years earlier showed
hemoglobin 11.4 g/dL, total leukocyte count 3100 per cu.mm and a
differential count of 49% polymorphs, 44% lymphocytes, 2%
eosinophils and 5% monocytes. Though his anti-tissue
transglutaminase antibody (IgA-tTG) was negative and duodenal biopsy
showed mild stunting of villi, he was put on gluten free diet (GFD)
as his IgA-antigliadin antibody was positive (32.4 U/mL, cut-off:
17). Despite strict adherence to GFD, the child did not show much
response and was further investigated at our institution. His
absolute neutrophil count was 840 per cu.mm with normal hemoglobin
and platelets. Serum amylase, lipase and blood sugar were within
normal limits. Stool examination showed high fecal fat excretion
(6.3 g/day after fat loading with 50 g butter) and Sudan stain
revealed 22 droplets of fat per high power field (normal up to 5
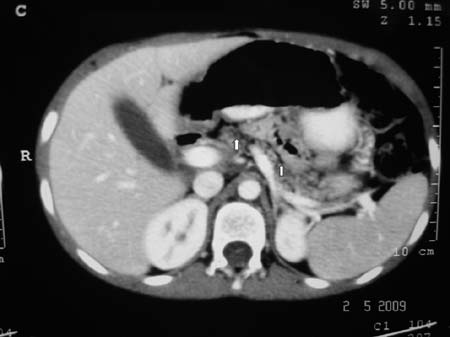
droplets). CT scan abdomen (Fig. 1) showed generalized
fatty replacement of pancreas without significant dilatation of main
pancreatic duct. Sweat chloride was 31 mEq/L (normal <40 mEq/L). He
had normal liver function tests, thyroid profile, serum
immunoglobulins, and negative antiendomysial antibody. Duodenal
biopsy was normal and bone marrow examination revealed mild marrow
hypoplasia.
 |
|
Fig. 1. Contrast enhanced CT scan
abdomen showing generalized fatty replacement of the
pancreas (white arrow).
|
A diagnosis of Shwachman-Diamond syndrome was
made and he was started on pancreatic enzyme and fat soluble vitamin
supplementations. On follow-up, steatorrhea settled and growth
improved (3.5 kg increase in weight and 6 cm increase in height
after 18 months). Intermittent neutropenia continued to occur on
follow-up but no evidence of serious infection except one episode of
Herpes labialis which was treated with acyclovir by the local
practitioner.
Discussion
When a patient presents with steatorrhea with
recurrent respiratory symptoms, the diagnosis first considered is
cystic fibrosis. As infections in our patient were not restricted to
lungs, the possibility of immune deficiency was also considered.
However, steatorrhea with intermittent nutropenia suggests the
diagnosis of SDS. Patients with SDS experience recurrent viral,
bacterial and fungal infections, including otitis media,
bronchopneumonia, osteomyelitis, skin infection and septicemia. The
quantitative and qualitative defects in neutrophils contribute to
these infections in SDS [2,6,7]. Failure to thrive is a common
manifestation because of malabsorption, recurrent infections and
metaphyseal dysostoses. The diagnostic criteria of SDS, as laid down
by Dror and Freedman, require documentation of exocrine pancreatic
dysfunction and characteristic hematological abnormalities [8].
Although not done in our case, genetic analysis can be performed for
confirmation. Shwachman-Bodian-Diamond syndrome (SBDS) gene is
located at chromosome 7q11 [6,9]. Ultrasonography shows normal sized
pancreas with increased echogenicity of the silhouette but CT and
MRI reveal lipomatosis of the pancreas with greater accuracy [6].
Common causes of pancreatic lipomatosis in children besides SDS are
cystic fibrosis, diabetes mellitus, obesity, Johanson-Blizzard
syndrome, Pearson’s marrow pancreas syndrome and agenesis or
hypoplasia of pancreas.
For gastrointestinal manifestations, the mainstay
of treatment is pancreatic enzyme therapy, medium-chain
triglyceride, and fat soluble vitamin supplements. With this
treatment, steatorrhea resolves and body weight increases but growth
is not generally accelerated [6]. Exocrine pancreatic function tends
to improve with time in almost half of the patients but
hematological problems deteriorate [6]. Our patient showed response
to enzyme supplements but continued to have problems related to
neutropenia. For the treatment of hematological abnormalities in
SDS, stem cell transplantation [10] and bone marrow transplantation
have been reported [3]. The projected median survival of patients
with SDS is 35 years and the main reason of untimely death is
hematological (bone-marrow failure, myelodysplastic syndrome and
acute myeloid leukaemia).
In conclusion, the diagnosis of SDS should be
strongly considered in any child who presents with steatorrhea,
growth retardation, and intermittent or persistent neutropenia.
Contributors: All authors contributed to
diagnosis and management of the case. RK drafted the article and, UP
and SKY critically reviewed it.
Funding: None; Competing interests:
None stated.
References
1. Shwachman H, Diamond LK, Oski FA, Khaw KT. The
syndrome of pancreatic insufficiency and bone marrow dysfunction. J
Pediatr. 1964;65:645-63.
2. Smith OP, Hann IM, Chessels JM, Reeves BR,
Milla P. Hematological abnormalities in Shwachman Diamond syndrome.
Br J Haematol. 1996;94:279-84.
3. Arseniev L, Diedrich H, Link H. Allogenic bone
marrow transplantation in a patient with Shwachman-Diamond syndrome.
Ann Hematol. 1996;72:83-4.
4. Kakkar N, Vasishta RK, Marwaha N, Marwaha RK.
Pathological case of the month. Arch Pediatr Adolesc Med.
2001;155:611-2.
5. Jha AK, Bansal D, Sharda S, Ray P, Steela L,
Varma N, et al. Shwachman-Diamond syndrome in India. Pediatr
Blood Cancer. 2012;58:479-80.
6. Hall GW, Dale P, Dodge JA. Shwachman Diamond
syndrome: UK perspective. Arch Dis Child. 2006;91: 512-24.
7. Burroughs L, Woolfrey A, Shimamura A.
Shwachman-Diamond syndrome: a review of the clinical presentation,
molecular pathogenesis, diagnosis and treatment. Hematol Oncol Clin
N Amer. 2009;23:233-48.
8. Dror Y, Freedman MH. Shwachman Diamond
Syndrome: A review. Br J Haematol. 2002; 118:701-13.
9. Boocock GR, Morrison JA, Popovic M, Richards
N, Ellis L, Durie PR, et al. Mutations in SBDS are associated
with Shwachman-Diamond syndrome. Nat Genet. 2003;33: 97-101.
10. Vibhakar R, Radhi M, Rumelhart S, Taatman D, Goldman F.
Successful unrelated umbilical cord blood transplantation in
children with Shwachman-Diamond syndrome. Bone Marrow Transplant.
2005;36:855-61.
|


