|
|
|
Indian Pediatr 2011;48: 729-731 |
 |
Satoyoshi Syndrome |
|
Debadatta Mukhopadhyay, Apurba Ghosh and Maya Mukhopadhyay
From the Institute of Child Health, Kolkata, West Bengal.
Correspondence to: Dr Maya Mukhopadhyay, Block GC - 109,
Sector 3, Salt Lake, Kolkata 700 106,
West Bengal, India.
Email: [email protected]
Received: April 05, 2010;
Initial review: April 28, 2010;
Accepted: June 21, 2010.
|
Satoyoshi syndrome is a rare autoimmune disease characterized by
alopecia, painful muscle spasms, diarrhea and secondary skeletal
changes. We report a 11-year old girl presenting with the typical
features of alopecia totalis, severe muscle spasm and skeletal
deformities.
Key words : Alopecia, India, Muscle spasm, Skeletal deformity.
|
|
Satoyoshi syndrome was first described by Satoyoshi and Yamada in
1967. It is common in Japanese population where it’s colloquial name
is komura-gaeri disease (komura implying calf and gaeri
implying spasm) [1]. Our patient presented with the classical
features of alopecia totalis, painful muscle spasms and skeletal
anomalies but did not show any evidence of endocrinopathy.
Case Report
This 11 year old girl, presented with
progressively increasing alopecia for last 5 years and painful
recurrent muscle spasms for last 3 months. These intense, painful
muscle spasms often occurred in the thigh, neck, around the knees,
and occasionally jaw spasms, and abdominal spasms simulating a
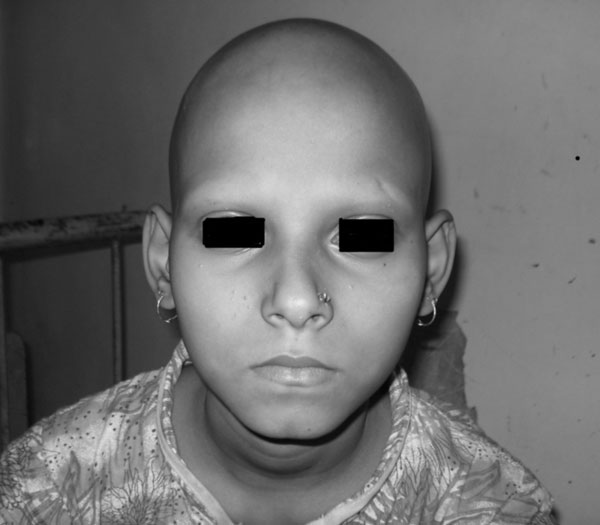
visible ill-defined swelling over abdomen. Loss of hair over scalp,
eyebrows (Fig. 1) and general body surface was alarming
to the parents. In addition, there was weight loss and deformity of
knees and lower limb resulting in movement restriction and limping.
Perinatal history and development were normal and there was no
history suggestive of neuroregression. School performance was
average, although the severe muscle cramps caused frequent
absenteeism.
 |
|
Fig. 1 Face showing complete loss of
hair from scalp and eyebrows. |
At 11 years, she weighed 18 kg (below 3rd
percentile), height was 131cm (at the 5th percentile), and head
circumference was normal. No secondary sexual characteristics had
appeared. There was total alopecia, generalized wasting, pallor,
bowing of lower limbs with genu valgum and pes planus. There was
normal muscle tone and grade 5 power in all muscles and no muscle
tenderness (except for episodes of spasm). Jerks were normal
bilaterally in both upper and lower limbs and there was no evidence
of any neurodeficit. Other systemic examination also did not reveal
any abnormality.
Laboratory evaluation revealed microcytic
hypo-chromic anemia and a normal blood count, liver and kidney
function tests. Bone marrow examination showed decreased iron stores.
Chest X-ray and USG were normal. Serum calcium, phosphate,
alkaline phosphate, CPK were normal. Endocrinal evaluation including
thyroid function tests, parathormone, growth hormone, follicle
stimulating hormone, leutenizing hormone were within normal limits.
Blood sugar and ANA levels were normal. Nerve conduction studies and
electromyography done during the spasm free periods were normal with
no spontaneous discharge at rest, normal motor unit potential and
normal interference pattern.
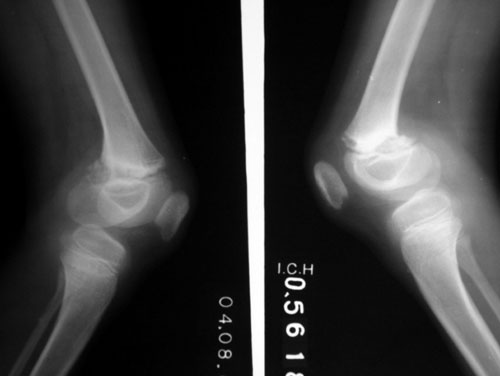
Skeletal survey revealed multiple defects: (i)
narrowing of ends of clavicle and terminal phalanges suggestive of
acroosteolysis (ii) irregular sclerotic distal femoral
metaphyses (Fig. 2), (iii) left sided genu varus,
right sided genu valgum; (iv) and delayed bone age.
 |
|
Fig. 2 Irregular sclerotic distal
femoral metaphyses. |
The child was treated with oral phenytoin (50 mg
twice daily), prednisolone (20 mg twice daily), Vitamin D and calcium
supplements. On follow up after 3 weeks, her spasms had significantly
improved but alopecia did not resolve. She is now spasm free but on
follow up and under consideration for methotrexate therapy.
Discussion
This multisystem disease occurs more commonly in
females, with mean age of onset around 10 years (range 6 to 15
years). Etiology is unknown and there is no established genetic
pattern yet described [2]. It is speculated to be a sporadic disease
of autoimmune origin [3, 4]. The autoimmune basis of the disease is
because of the improvement with steroids, its association with other
autoimmune diseases, deposition of immune complexes in the muscles,
and in few cases, a positive anti-nuclear antibody [3,5]. Studies
have also demonstrated antibodies against brain and gastrointestinal
tissue [6].
The characteristic painful intermittent muscle
spasms are progressive, frequently severe enough to cause abnormal
posturing of the limbs, and lasting several minutes. It may progress
to involve the limb girdle muscles and also the temporalis and
masseters and rarely may interfere with speech and respiration. The
diarrhea may lead to carbohydrate malabsorption. The endocrinopathy
usually manifests as amenorrhea or as hypoplastic uterus [3].
The unique feature of Satayoshi’s syndrome are the
myriad skeletal abnormalities presumed to be due to recurrent
vigorous muscle spasms causing repeated injuries to the growth
plates, epiphyses, and tendon attachments in the growing skeleton
[7]. Severe muscle spasms may respond to intravenous calcium
gluconate, dantrolene sodium, quinine, procainamide and phenytoin
[8]. Refractory spasms may be treated with botulinum toxin [9]. In
those patients with severe side effects to long term glucocorticoids,
a safer alternative is frequent pulse therapy with intravenous immune
globulin [10].
Contributors: DM: data compilation and
manuscript drafting; AG: guidance for manuscript writing and design;
MM: guidance for manuscript writing and design.
Funding : None.
Competing interests: None stated.
References
1. Satoyoshi E, Yamada K. Recurrent muscle spasms
of central origin. A report of two cases. Arch Neurol.
1967;16:254-64.
2. Salah Uddina ABM., Waltersabc AS, Alie A,
Brannanad TA. Unilateral presentation of ‘Satoyoshi syndrome’.
Parkinsonism Related Disorders. 2002; 8:211-3.
3. Ashalatha R, Kishore A, Sharada C, Nair MD.
Satoyoshi syndrome. Neurol India. 2004;52:94-5.
4. Drost G, Verrips A, Van Engelen BGM, Stegeman
DF, Zwarts MJ. Involuntary painful muscle spasms in Satoyoshi
syndrome: a surface electromyographic study. Mov Disord.
2006;11:2015-8.
5. Asherson RA, Giampaolo D, Strimling. A case of
adult-onset Satoyoshi syndrome with gastric ulceration and
eosinophilic enteritis. Nature Clin Pract Rheumatol.
2008;4:439-44.
6. Matsuura E, Matsuyama W, Sameshima T, Arimura
K. Satoyoshi syndrome has antibody against brain and gastrointestinal
tissue. Muscle Nerve. 2007;36:400–3.
7. Ikegawa S, Nagano A, Satoyoshi E.
Skeletal abnormalities in Satoyoshi’s syndrome: a radiographic study
of eight cases. Skeletal Radiol. 1993;22:321-4.
8. Harati Y, Kolimas RJ. Muscle cramps, stiffness
and myalgia. In: Jankovic J, Tolosa E, eds. Parkinson’s
Disease and Movement Disorders. USA: Williams and Wilkins. 1998. p.
796-8.
9. Merello M, Garcia H, Nogues M, Leiguarda R.
Masticatory muscle spasms in a non-Japanese patient with Satoyoshi
syndrome successfully treated with botulinum toxin. Mov Disord.
1994;9:104-5.
10. Arita J, Hamano S, Nara T, Maekawa K.
Intravenous gammaglobulin therapy in Satoyoshi syndrome. Brain Dev.
1996;18:409-11.
|
|
|
 |
|
