|
|
|
Indian Pediatr 2011;48: 727-729 |
 |
Dysmyelination of the Cerebral White matter
with Microdeletion at 6p25 |
|
Seema Kapoor, *Sharmila Banerjee Mukherjee,
†Daraius
Shroff and Ritu Arora
From the Departments of Pediatrics: Maulana Azad Medical
College, and *Lady Hardinge Medical College, New Delhi; and †Department of
Ophthalmology, Guru Nanak Eye Centre, Maulana Azad Medical College, New
Delhi.
Correspondence to: Dr Seema Kapoor, M439, Guruharkrishan
Nagar, Paschim Vihar, New Delhi 110 087.
Email: drseemakapoor@gmail.com
Received: January 19, 2010;
Initial review: March 25, 2010;
Accepted: June 1, 2010.
|
A 6-year old boy presented with mental retardation, hypotonia, abnormal
facies, impaired hearing, protuberant eyes, visual impairment, short
stature, Axenfeld-Rieger anomaly, a bicuspid aortic valve, and bilateral
sensorineural deafness. CT scan of head suggested dysmyelination of the
subcortical and periventricular white matter. FISH revealed a
subtelomeric microdeletion encompassing both FOXC1 and FOXF2 loci within
6p25. Dysmyelination of the central nervous system has been infrequently
described earlier in patients with 6p25 deletion.
Key words: 6p25 microdeletion, Axenfeld-Rieger anomaly,
Dysmyelination.
|
|
Axenfeld-Rieger anomaly (ARA) is a spectrum of developmental anomalies of
the anterior chamber of the eye that predispose to glaucoma in childhood.
Axenfeld-Rieger syndrome (ARS) encompasses Axenfeld (anterior segment
defect) and Rieger anomaly (anterior segment with iris defects) with
systemic manifestations, including Rieger syndrome (ocular abnormalities,
umbilical hernia and dental anomalies) and a variety of multiple
congenital anomaly syndromes [1]. ARS is genetically heterogenous but an
increasingly recognized cause of ARS is 6p deletion, which gives rise to
an identifiable pattern of malformations, characterized by AR anomaly,
sensorineural hearing loss, cardiac, cerebral and craniofacial anomalies
[2-4]. Dysmyelination of the white matter has been infrequently described
with ARS [3, 4].
Case Report
A six-year old, the third child of healthy unrelated
parents and born at term following an uncomplicated pregnancy. Immediate
postnatal complications or feeding difficulty, had delayed
acquisition of developmental milestones, particularly in the domains of
gross motor and language. He presented with progressive deterioration of
vision, hearing impairment, proportionate short stature and mental
retardation. There was history of delayed descent of both testes noticed
at 5 years. His parents were of average intelligence, normal stature and
had no ocular or facial abnormalities.
On examination, he had proportionate short stature (100
cm, 3-4 SD below 50th centile), weight of 15 kg, and a head circumference
of 50 cm. The child had a broad, high forehead, shallow orbits, proptosis,
hypertelorism, down-slanting palpebral fissures, a flat nasal bridge, a
short upturned nose, maxillary hypoplasia, a long featureless philtrum,
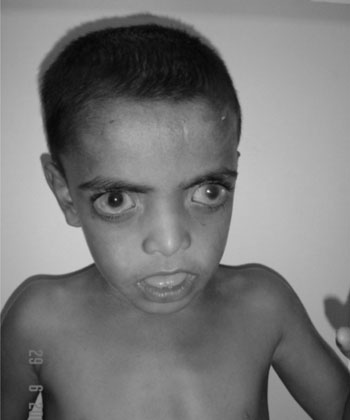
thin upper vermillion border, open mouth and protuberant tongue (Fig.
1). Generalized joint laxity was present. The umbilicus and
external genitalia were normal. There was a grade III ejection systolic
murmur at the aortic area that radiated to the neck. Central nervous
system examination revealed hypotonia with normal reflexes with normal
muscle power. The cranial nerve examination was normal with no cerebellar
signs. He walked with a slightly broad based gait. The intelligence
quotient (IQ) was 45 (WISC scale).
 |
|
Fig. 1 Photograph of the child showing
hypertelorism, featureless philthrum, and open mouth. |
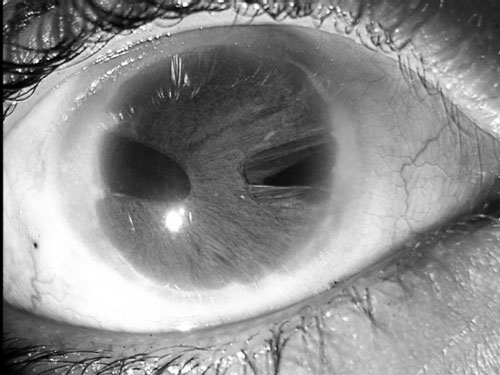
Slit lamp examination showed bilateral posterior
embryotoxon. In the right eye, there was corectopia, pseudopolycoria
(full-thickness colobomas of the iris which may appear to be multiple
pupils) and generalized extensive areas of iris stromal hypoplasia with
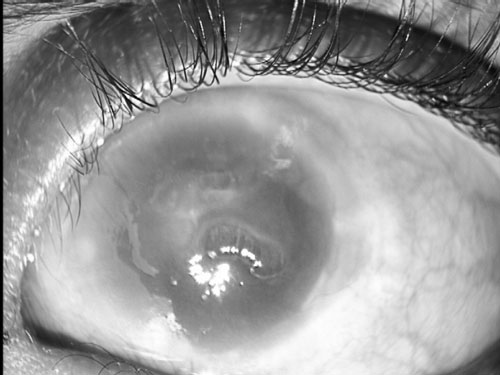
normal corneal clarity (Fig. 2a). The left cornea was
hazy with a central opacity and severe edema with epithelial bullae (Fig.
2b). Corectopia was identified but delineation of anterior
chamber was limited by the degree of opacity. The right and left
intraocular tension was 26 mm Hg and 30 mm Hg, respectively. Visual acuity
was 20/200 on the right with only hand movements close to face detected
with the left eye. Radioimaging of the skeletal system revealed
generalized osteopenia. The spine and skull were normal. The thyroid
function tests were normal. Echocardiography revealed a mildly stenotic
bicuspid aortic valve. The CT scan of the brain revealed dysmyelination of
the subcortical and periventricular white matter. Renal ultrasound was
normal. The BERA demonstrated bilateral moderate sensorineural deafness.
|
(a) |
(b) |
|
Fig.2 Right eye showing corectopia(a) and
left eye showing hazy cornea with a central corneal opacity and
severe corneal edema with epithelial bullae (b). |
A high resolution (550 band) GTL karyotype from
peripheral lymphocytes was 46, XY, with a normal banding pattern. FISH
analysis was performed using Clone dJ668J24(FOXF2) and Clone
dJ118B18(FOXC1), both of which hybridize to 6p subtelomere (Vysis, Des
Plaines, IL) demonstrating deletion of both these p-arm sub-telomere loci,
from one homologue of chromosome 6. This deletion extended proximally, to
include the FOXC1 and FOXF2 loci within 6p25. No copies of the deleted
loci were found on any other elements of the karyotype. The conventional
karyotype of the parents were normal; FISH analysis could not be performed
for economic reasons.
Discussion
Fifteen mutations have been identified in the FOXC1
gene to date. Mutations in FOXF2 and FOXQ1 have also been reported. FOXF2
is expressed in the anterior segment of the eye, inner ear and pia mater
indicating that it may contribute to the ocular, auditory and neurological
phenotypes of this syndrome [5]. Lehmann, et al. [6] detected both
interstitial duplications and deletions of 6p25 in Axenfeld-Rieger
syndrome. All the rearrange-ments encompassed FOXC1. The consistent
findings in the 6p25 deletions are developmental delay/mental retardation,
hypotonia, craniofacial, ophthalmologic and cardiac anomalies. Genital,
palatal, skeletal (clino-dactyly), and central nervous system (CNS)
manifestations are more variable. The CNS anomalies which have been
reported are hydrocephalus, pachygyria, cavum septum pellucidum and
hypoplasia of the cerebellum, brainstem, and corpus callosum.
The Axenfeld-Rieger malformation is seen in several
other syndromes. Moog, et al.[7] reported two sisters who presented
with Axenfeld-Rieger anomaly, hydrocephalus, leptomeningeal calcifications
and mild mental retardation. Another entity with Reiger anomaly is SHORT
syndrome [8].
Variable developmental defects have been reported from
near normal cognition to severe developmental delay. Dysmyelination of the
periventricular white matter has been infrequently reported and adds to
the causes of delay seen in 6p25 phenotype, and may explain the
developmental delay seen in our case. Recent studies in mice have reported
the role of FOXC1 gene in cortical development in which hypomorphic
mutations affect all the three meningeal layers with severe consequences
in the development of the skull and underlying brain tissue, thereby
explaining the cortical dysgenesis seen in our case. Since the breakpoint
encompasses this gene the phenotype can be explained [9].
The constellation of clinical features reported and a
distinctive phenotype should prompt the clinician to investigate for a
6p25 deletion. Submicroscopic 6p deletion appears to be a recognizable
clinical phenotype, and this region should be thoroughly investigated with
FISH probes, including at least a subtelomeric 6p probe and a probe
covering FOXC1, for patients presenting with these features.
Acknowledgment: We thank Dr Ken Maclean, Clinical
Geneticist, Gutav Nossal NH & MRC scholar. Victor Chang Cardiac Research
Institute, Australia and Dr Gregory Peters Luke St.Heaps, of the
Cytogenetics Dept, Children’s Hospital at Westmead, Sydney, Australia, for
carrying out all of the FISH studies described herein.
Contributors: SK made the diagnosis, was
responsible for literature search; SBD and DS prepared the manuscript; SK
and RA critically evaluated the paper and gave the final approval.
Funding: None.
Competing interests: None stated.
References
1. Fitch N, Kaback M.. The Axenfeld syndrome and the
Rieger syndrome. J Med Genet. 1978:15:30-4.
2. Maclean K, Smith J, Heaps L, Chia N, Williams R,
Peters GB, et al. Axenfeld-Rieger malformation and distinctive
facial features: clues to a recognizable 6p25 microdeletion syndrome.
Am J Med Gent. 2005;132A:381-5.
3. Kniestedt C, Taralczak M, Thiel MA, Stuermer J,
Baumer A, Gloor BP. A novel PITX2 mutation and polymorphism in a
5-generation family with Axenfeld- Reiger anomaly and coexisting Fuch’s
endothelial dystrophy. Opthalmology. 2006;113:1791.E 1-8.
4. Kaestner KH, Knochel W, Martinez DE. Unified
nomenclature for the winged helix/ forkhead transcription factors. Genes
Dev. 2000:14: 142-6.
5. Law CJ, Fisher AM, Temple IK. Distal 6p deletion
syndrome: A report of a case with anterior chamber anomaly and review of
published reports. J Med Genet. 1998;35:685-9.
6. Lehmann OJ, Ebenezer ND, Ekong R, Ocaka L, Mungall
AJ, Fraser S, et al. Ocular developmental abnormalities and
glaucoma associated with interstitial 6p25 duplications and deletions.
Invest Ophthal Vis Science. 2000;43:1843-9.
7. Moog U, Bleeker-Wagemakers EM, Crobach P, Vles JSH,
Schrander-Stumpel CTRM. Sibs with Axenfeld-Rieger anomaly, hydrocephalus
and leptomeningeal calcifications: A new autosomal recessive syndrome? Am
J Med Genet. 1998;78:263-6.
8. Brooks JK, Coccaro PJ Jr, Zarbin MA. The Rieger
anomaly concomitant with multiple dental, craniofacial, and somatic
midline anomalies and short stature. Oral Surg Oral Med Oral Path.
1989;68:717-24.
9. Zarbalis K, Siegenthaler JA, Choe Y, May SR,
Peterson AS, Pleasure SJ. Cortical dysplasia and skull defects in mice
with a Foxc1 allele reveal the role of meningeal differentiation in
regulating cortical development. Proc Natl Acad Sci USA.
2007;104:14002-7.
|
|
|
 |
|
