|
|
|
Indian Pediatr 2009;46: 804-806 |
 |
Early Myoclonic Encephalopathy |
|
Mahesh Kamate, Niranjana Mahantshetti and Vivek Chetal
From the Department of Pediatrics, KLE University’s J N
Medical College, Belgaum, Karnataka State, India.
Correspondence to: Dr Mahesh Kamate, Assistant Professor
of Pediatrics and In-charge, Child Development Clinic, KLE University’s J
N Medical College, Belgaum, Karnataka, India.
Email: [email protected]
Manuscript received: July 21, 2008;
Initial review: August 18, 2008;
Accepted: November 14, 2008.
|
|
Abstract
Early myoclonic encephalopathy (EME) is a rare
malignant epileptic syndrome. The erratic myoclonus with or without
focal motor seizures, onset before 3 months of age, and persistent
suppression-burst pattern in electroencephalograph (EEG) are accepted as
the diagnostic criteria for EME. We report an 11 month-old infant with
EME which was secondary to non-ketotic hyperglycinemia.
Keywords: Early myoclonic encephalopathy, Epilepsy, Infant,
Non-ketotic hyperglycinemia, Suppression-burst patttern.
|
|
E
arly myoclonic encephalopathy (EME)
is one of the two recognized epileptic encephalopathies which are seen in
early infancy, the other being early infantile epileptic encephalopathy (EIEE
or Ohtahara syndrome). EME is characterized by characteristic suppression
burst (SB) on electroencephalogram (EEG). It is a malignant epilepsy
syndrome(1).
According to the International Classification of
Epilepsies and Epileptic Syndromes (ILAE, 1989), EME is categorized as
age-related, generalized symptomatic epilepsy of non-specific etiology(2).
EME is clinically characterized by the onset of erratic or fragmentary
myoclonus (usually involving the face or extremities), massive myoclonus,
other refractory partial seizures and marked neurologic
abnor-malities(2,3). We report an 11 month-old infant presenting with EME,
who on metabolic work was found to have non-ketotic hyperglycinemia (NKH).
Case Report
An 11 month-old infant, born to second-degree
consanguineously married couple, presented to us with recurrent seizures,
developmental delay and recurrent respiratory tract infections. He was
born normally after an uncomplicated term pregnancy. After birth, baby was
hypotonic and had difficulty in sucking and swallowing. He developed
myoclonic and focal tonic seizures in the first week of life and was
started on phenobarbitone. Biochemical tests (including calcium, magnesium
and glucose) done at that time and MRI brain were normal. EEG done at 3
months showed SB pattern with multifocal spikes and sharp waves and by
then, child had developed erratic fragmentary myoclonia. Child was
receiving adequate doses of valproate and clobazam but the child continued
to have myoclonic seizures and frequent tonic seizures appeared by 4-5
months. Child did not attain any milestones expected for his age and
remained floppy.
He presented to us at 11 months with an episode of
bronchopneumonia which was managed with antibiotics. His weight, length,
and head circumference were within normal limits. He was lethargic and had
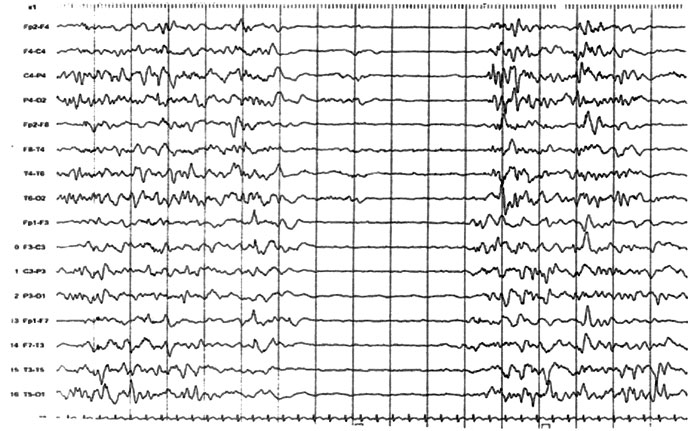
hypotonia with hyperreflexia. EEG was repeated and it showed SB pattern (Fig.1).
In view of erratic myoclonia, tonic seizures, severe developmental delay,
hypotonia and persistent SB pattern on EEG, a diagnosis of EME was
considered. Repeat MRI brain was normal. Tandem mass spectrometry of
blood, revealed high glycine levels (1068.48 µmol/L; normal 2-745 µmol/L)
suggesting NKH. Enzymatic analysis or mutational studies could not be done
and a diagnosis of EME secondary to NKH was made. We tried to control the
seizures with high-dose valproate, lamotrigine and pyridoxal phosphate.
Dextromethorphan (25 mg/kg/day) in combination with benzoate (500-750
mg/kg/day) was prescribed for NKH. However, seizures continued to persist
and there was no much improvement. Parents were counseled regarding the
outcome and recurrence risk.
 |
|
Fig.1 Sleep EEG showing suppression burst
pattern and poorly organized background. Bursts lasts for 1-5
seconds alternating 3-10 seconds suppression. |
Discussion
Early myoclonic encephalopathy is a rare epileptic
syndrome with onset nearly always in the first three months of life,
mostly within the neonatal period. The main ictal manifestations are
partial or fragmentary erratic myoclonus, massive myoclonus, partial motor
seizures and tonic infantile spasms(5). The usual and earliest seizure
type is fragmentary myoclonus and is regarded as an essential symptom in
EME. The closest differential diagnosis for EME is Ohtahara syndrome (OS)
which has an early onset, within a few months of birth, and frequent tonic
spasms with or without clustering, as the main seizure type. Unlike EME,
more than two third cases of OS evolve to West syndrome at 4-5 months of
age(5).
EEG shows SB pattern in which complex bursts of spikes,
sharp waves and slow waves are separated by episodes of flattening of the
tracing and localized discharges that resemble those of neonatal seizures.
The bursts last for 1-5 seconds and alternate with 3-10 seconds of
suppression. This SB pattern is more distinct during sleep, especially
deep sleep(4,5). EEG later evolves towards atypical hypsarrhythmia or
multifocal paroxysms at 3-5 months of age. However, in most cases, this
phase is transient, and a return to the SB pattern is observed. The SB
pattern primarily reflects a diffuse structural or junctional disturbance
of gray matter connectivity. In OS, the most characteristic EEG feature is
also SB pattern, but this is consistently seen during both awake and sleep
states and later evolves to hypsarrhythmia in many cases in first six
months of life. A persistent SB pattern beyond 6-8months of age on EEG is
pointer towards EME(1,3,4).
There are no clear guidelines for treatment of seizures
in EME. Conventional antiepileptic drugs, ACTH, corticosteroids and
pyridoxine are ineffective in controlling the seizures(3). Even the
alternative strategies for epilepsy like ACTH, ketogenic diet and
zonisamide were found to be more beneficial in OS than EME(6). The
prognosis is poor as more than 50% patients die before one year of age and
the remaining enter a vegetative state. In patients with NKH, oral
administration of ketamine (8 mg/kg/day, in four divided doses),
tryptophan (100-150 mg/kg/day) and dextromethorphan (5-35 mg/kg/day) in
combination with benzoate (500-750 mg/kg/day) have brought about only
partial improvement of neurological symptoms and EEG findings(7).
Non ketotic hyperglycinemia, being an autosomal
recessive condition, there is 25% chance of recurrence in the next
pregnancy. This is important for genetic counseling and prenatal
diagnosis.
Acknowledgment
Dr Rita Christopher from the Department of
Neurochemistry, National Institute of Mental Health and Neurosciences,
Bangalore for the Tandem Mass Spectrometry analysis.
Contributors: MK diagnosed and managed the case. MK
drafted the article and will act a guarantor of the manuscript. VK did
literature search and helped in drafting. NM revised the paper critically.
Funding: None.
Competing interests: None stated.
References
1. Chen PT, Young C, Lee WT, Wang PJ, Peng SS, Shen YZ.
Early epileptic encephalopathy with suppression burst
electroencephalographic pattern- an analysis of eight Taiwanase patients.
Brain Dev 2001; 23: 715-720.
2. Commission on Classification and Terminology of
International League Against Epilepsy: Proposal for revised Classification
of Epilepsies and Epileptic syndromes. Epilepsia 1989; 30: 389-399.
3. Ohtahara S, Ohtsuka Y, Oka E. Epileptic
encephalopathies in early infancy. Indian J Pediatr 1997; 64: 603-612.
4. Ozyurek H, Turanli G, Aliefendioglu D, Coskun T.
Repetitive EEG recordings are necessary for the diagnosis of early
myoclonic encephalopathy. Neurol India 2005; 53: 235-237.
5. Ohtahara S, Ohtsuka Y, Erba G. Early pileptic
encephalopathy with suppression burst. In: Engel J Jr, Pedley TA,
Eds. Epilepsy - A comprehensive Textbook. Vol III. Philadelphia:
Lippincott-Raven Publishers; 1997.
6. Ohno M, Shimotsuji Y, Abe J, Shimada M, Tamiya H.
Zonisamide treatment of early infantile epileptic encephalopathy. Pediatr
Neurol 2000; 23: 341-344.
7. Sehgal V, Ramji S. Nonketotic hyperglycinemia in a neonate. Indian
Pediatr 1998; 35: 278-281.
|
|
|
 |
|
