|
|
Case Report Indian Pediatrics 2008; 45:780-782 |
|||||||
|
Bartsocas-Papas Syndrome |
|||||||
|
Koumudi Godbole From the Departments of Genetic Medicine, Pathology,
and Obstetrics and Gynecology, Deenanath Mangeshkar Correspondence to: Dr. Koumudi Godbole
Consultant Clinical Geneticist, Dept of Genetic Medicine, Deenanath Manuscript received: August 27, 2007; Initial review
completed: October 22, 2007; Abstract
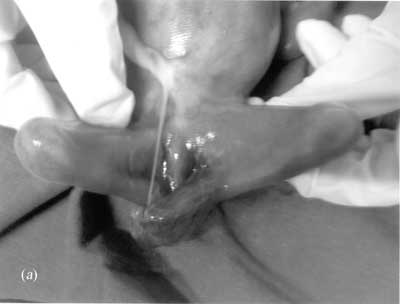
Bartsocas-Papas syndrome (BPS) MIM 263650) is a severe and rare autosomal recessive syndrome characterized by popliteal pterygium / webbing, oligo-syndactyly, genital anomalies and a typical face with short palpebral fissures, ankyloblepharon, hypoplastic nose, oro-facial clefts and small mouth(1). Most of the reported cases either die in-utero or in the neonatal period, although occasional survival has been reported with aggressive management(2-4). We report a 3rd degree consanguineous Indian family belonging to Mahadeo Koli community with three consecutive pregnancies affected by Bartsocas-Papas syndrome. Case Report First pregnancy: The first pregnancy was reportedly complicated by moderate to severe oligohydramnios and decreased fetal movements. A female baby with low birth weight was born at term by cesarean section and had multiple anomalies including facial dysmorphism and skin folds extending across the limbs. This baby also had fused fingers and the nose appeared particularly deformed. The baby expired at about 8 weeks while on supportive therapy. Karyotype was reportedly normal. No medical documents or photographs were available for review. Second pregnancy: The second pregnancy was uneventful up to 19 weeks with normal nuchal translucency at 12 weeks and low risk triple marker screen at 16 weeks of pregnancy. The mother was on nifedipine for treatment of mild hypertension of pre-pregnancy onset and used salbutamol inhaler for about 2 weeks in the first five months of pregnancy. Although no structural abnormality or pterygia were detected on prenatal ultrasound, progressive reduction of amniotic fluid volume and absent fetal movements was noted. The pregnancy was terminated at 21 weeks and the fetus was found to have features typical of BPS including intrauterine growth retardation, joint contractures with pterygia extending from axilla to chest, between the elbows and popliteal area of lower limbs. There were bands of fibrous tissue extending from abdomen to feet of the fused lower limbs and peri-anal area. The feet and hands were like nubbins without definable toes or fingers. The external genitals were unidentifiable. Facial dysmorphism included extreme micrognathia, retracted eyelids, facial cleft, hypertelorism, depressed nasal bridge and low set ears (Fig. 1).
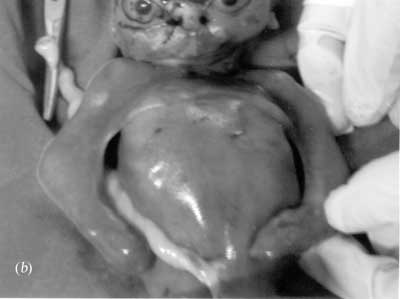
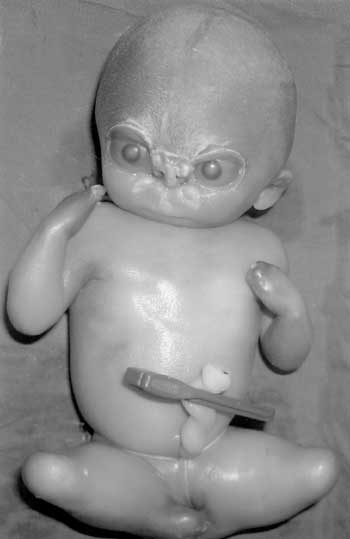
The fetal karyotype obtained after termination was normal, 46 XY. The placental histopathology was normal without any evidence of placental dysfunction or infection. Muscle and brain histopathology and fetal X-rays were normal. Third pregnancy: This pregnancy was also terminated at 20 weeks in view of absent fetal movements, progressive reduction in amniotic fluid volume and possible recurrence of BPS. The fetus was detected to have almost identical features as that of the second fetus with length of 21 cm and head circumference of 24 cm. The popliteal pterygium was very extensive extending from the thigh to the ankle bilaterally in addition to the pterygia in the axilla and cubital fossa. There was oligo-syndactyly, ankyloblepharon with hypertelorism and malformed nose with cleft palate. The external genitalia were indefinable (Fig. 2).
Discussion BPS is a subgroup of multiple pterygium syndromes and falls into the subcategory of autosomal recessive lethal variants as described by Hall, et al.(5). The fetuses described in this report had clinical features consistent with BPS including popliteal pterygium, and other typical facial features including clefting and ankyloblepharon, oligo-syndactyly of fingers and absence of bony fusions and nuchal edema. Neither fetal autopsy nor radiology revealed any internal or skeletal anomalies. Our case did not have polyhydramnios as reported in some cases previously. Morgan, et al.(6) reported CHRNG (embryonal subunit of the acetylcholine receptor) mutations in both lethal and non-lethal variant (Escobar syndrome) of multiple pterygium syndromes but not specifically in BPS families. Shanske, et al.(7) have reported absence of IRF6 (Interferon regulatory factor 6) gene mutations in a family with two sibs affected with BPS(7). No specific developmental pathological explanation is available to explain all the findings seen in BPS, which might help in candidate gene studies. Nosological differences between the BPS and other major varieties of lethal popliteal pterygium syndromes have been previously described by Papadia and Longo(8). In absence of a specific genetic diagnosis, a careful follow up by ultrasound remains the current method of prenatal diagnosis in Bartsocas Papas syndrome. Acknowledgement We thank Dr. Manali Ketkar, referring Obstetrician and Gynecologist for involving us in the patient’s care. Contributors: KG and GG were involved in patient management, reviewing the literature and writing the manuscript. VB was involved in pathology examination and reviewing the literature. KG will stand guarantor. Funding: None. Competing interest: None stated.
| |||||||
![]()