|
|
Case Reports Indian Pediatrics 2008; 45:777-779 |
|||||
|
Biotinidase Deficiency |
|||||
|
Ramdas Dahiphale Shreepal Jain Mukesh Agrawal From the Department of Pediatrics, Topiwala National Medical College & B.Y.L.Nair Hospital, Mumbai, India. Correspondence to: Ramdas Dahiphale, Department of Pediatrics, Topiwala National Medical College & B.Y.L.Nair Hospital, Mumbai 400 008, India. E-mail: [email protected] Manuscript received:November 20, 2007; Initial review
completed: January 24, 2008; Abstract
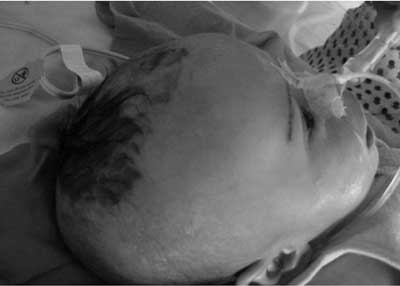
Biotinidase deficiency, first described in 1983, can be profound (<10% enzyme level) or partial (10-30% enzyme level). Clinical manifestations include neurological, dermatological, immunological, and ophthalmological abnormalities. We report a case of profound biotinidase deficiency(1). Case Report A 3-month old boy, born of nonconsanguineous marriage, presented with sudden onset of breathlessness, lethargy and refusal of feed for twelve hours. He had one episode of multiple seizures at 2 months of age. MRI brain at that time revealed ischemic changes in bilateral parietal lobe and subcortical matter. He was treated symptomatically, followed by long-term anticonvulsant therapy with phenobarbitone. On admission, he had hypothermia, tachycardia, tachypnea with grunting and signs of dehydration. Skin showed an erythematous rash around neck, back and perineal region. He had conjunctivitis, alopecia and seborrheic dermatitis since birth (Fig. 1). The child was drowsy, responding to only painful stimuli. Other systemic examination was normal. Laboratory investigations revealed normal hemogram, liver functions, serum ammonia and serum electrolytes. Blood gas analysis showed persistent severe metabolic acidosis, refractory to therapy. Baby also had persistent ketonuria. CSF was normal.
After admission, baby was treated in intensive care but deteriorated neurologically with progressive coma and required ventilatory support. Subsequent review of previous MRI suggested vasogenic edema in bilateral corona radiata. In view of poor therapeutic response and suspicious MRI reports, he was investigated further for a metabolic disorder. Plasma and urine aminoacidogram was normal. Tandem mass spectrometry revealed increased C5OH levels, suggestive of holocarboxylase deficiency. Specific enzyme assay showed deficient biotinidase activity of 0.2 nmol/min/mL (normal >5nmol/min/mL). He was started on oral biotin (10mg/day). The child improved dramatically within few days with normalization of sensorium and blood gas reports, control of seizures, and disappearance of skin lesions. He was discharged after 20 days on biotin supplements and presently doing well after 6 months. Discussion Biotinidase deficiency is a rare metabolic disorder with an estimated incidence of 1:61067 population, though severe or profound disease is much rare (1: 137401 population)(2). Only two reports of biotinidase deficiency could be traced in Indian literature, of which one was detected on routine neonatal screening(3-5). Clinical presentation depends on the severity of enzymatic defect. Profound defects usually manifest between 3-6 months of age with - (a) neurological manifestations (seizures, hypotonia, and develop-mental delay); (b) skin manifestations (eczematous skin rash, seborrheic dermatitis, alopecia); and (c) respiratory problems (hyperventilation, laryngeal stridor and apnea). Conjunctivitis is common. Older children and adolescents may exhibit limb weakness, neurosensory hearing loss and eye problems e.g. optic atrophy and scotomas. Present case did not have ocular or auditory problems. Many symptomatic children with biotinidase deficiency exhibit various neuroimaging abnormalities e.g. cerebral edema, attenuated white matter signal, cerebral atrophy, and compensatory ventricular enlargement. Neuroimaging features may improve or become normal after biotin treatment(6). Candidiasis is common, which may be related to immunological dysfunction following accumulation of toxic metabolites or biotin deficiency itself. Children with partial biotinidase deficiency (10-30% activity) may present with milder symptoms, especially during stress e.g., infection. Most cases with Biotinidase deficiency exhibit metabolic ketolactic acidosis, organic aciduria, and mild hyperammonemia. However the absence of organic aciduria or metabolic ketoacidosis does not exclude the diagnosis in a symptomatic child. Biotinidase deficiency may be detected on newborn screening(7) or by prenatal molecular diagnosis for mutations, recommended in case of previous affected child in the family. Recently, molecular genetic tests have been used for detection of carrier state(8). Apart from common neonatal conditions e.g. sepsis, meningitis or epilepsy, biotinidase deficiency may be confused with holocarboxylase deficiency previously called early-onset or infantile multiple or combined carboxylase deficiency. In present case, a negative sepsis workup, paucity of clinico-radiological signs despite marked breathlessness, skin changes, severe metabolic acidosis and ketonuria led us to suspect biotinidase deficiency, which was confirmed on enzyme analysis.
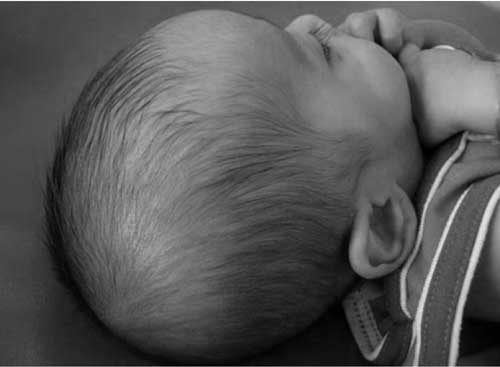
Biotinidase deficiency needs life-long biotin supplements upto 40mg/day. Raw eggs, containing avidin that binds biotin, must be avoided. Present case responded well to therapy (Fig 2). The prognosis is excellent though some features e.g. optic atrophy, hearing loss or developmental delay may not be reversible and should be addressed with periodic evaluations and relevant interventions(9). Contributors: RD managed the patient and drafted the manuscript. SJ reviewed the literature, MA supervised patient care, edited the paper and will act as guarantor. Funding: None. Competing interests: None stated. .
|
![]()