|
|
Images in Clinical Practice Indian Pediatrics 2002; 39:876-878 |
|
Apert Syndrome |
|
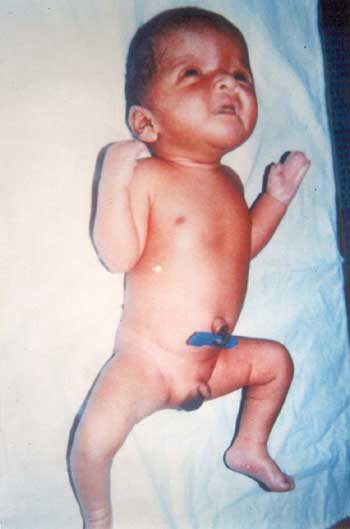
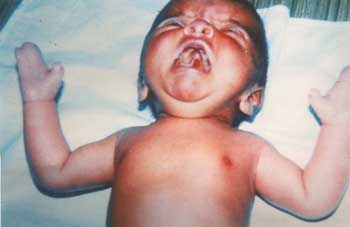
A 7 days old male neonate from an orphanage, presented with loose stools. On examination, he had flattened occiput, prominent coronal suture, hypertelorism, depressed nasal bridge, antimongoloid slant, maxillary hypoplasia and small nose (Fig.1). Examination of oral cavity revealed cleft of soft palate (Fig. 2). Syndactyly of both hands with complete fusion of 2nd, 3rd, 4th and 5th fingers with broad distal phalanx of thumb and syndactyly of all toes were other prominent features. The fused fingers and toes had separate nails. There were no other skeletal deformities and his fundus examination was
normal. Cardiovascular system examination revealed a systolic murmur over the pulmonary area. His neurosonogram and ultrasonogram abdomen were normal but echo cardiogram showed an atrial septal defect. Apert syndrome was first described by Eugene Apert in 1906 as a triad of craniosynostosis, syndactyly and maxillary hypoplasia. It is an autosomal dominant disorder but majority of cases are sporadic and associated with older paternal age. Apert Syndrome is characterized by irregular craniosynostosis, mid facial hypoplasia, syndactyly, broad distal phalanx of thumb and big toe. Mutations in the fibroblast growth factor receptor 2 gene which maps to chromosome 10q 25 – 10q 26 cause Apert Syndrome. Majority of patients have mental retardation. Agenesis of corpus callosum, progressive hydrocephalus and hippocampal abnormalities can occur in children with Apert Syndrome. Early surgery for craniosynostosis is indicated when there is evidence of increased intracranial pressure. When the thumb is immobilized, early surgerymm should be done to facilitate pincer grasp. A.M. Vijayalakshmi, Ajay Menon, Department of Pediatrics P.S.G. Institute of Medical Science & Research, Peelamedu, Coimbatore 641 004, Tamil Nadu, India
|
![]()