|
|
|
Indian Pediatr 2020;57:
975-976 |
 |
Proximal Limb Girdle Weakness, Joint
Hyperlaxity, and preserved Deep Tendon Reflexes: A Distinctive
Phenotype
|
|
Priyanka Madaan and Lokesh Saini *
Pediatric Neurology Unit, Department of Pediatrics,
Postgraduate Institute of Medical Education and Research, Chandigarh,
India.
Email: [email protected]
|
|
A 9-year-old girl presented with mild motor delay and progressive
proximal limb-girdle weakness. Socio-cognitive milestones were normally
attained. Examination revealed normal head size and intellectual
functioning, proximal limb girdle weakness, mildly prominent calves, and
preserved deep tendon jerks (including both ankles). She had hyperlaxity
of finger joints and both elbow joints (Beighton score 4/9). She also
had polyminimyoclonus. Creatine kinase levels were elevated (790 IU/L)
while electrocardiogram revealed tremor (Fig. 1). Nerve
conduction studies revealed motor axonal loss with sensory sparing while
electromyography (EMG) was suggestive of abnormal spontaneous activity
(fibrillations and fasciculations) signifying active denervation. She
was not cooperative for voluntary EMG assessment. Multiplex
ligation-dependent probe amplification (MLPA) revealed homozygous
deletion of exon 7 and 8 of SMN1 gene confirming the diagnosis of
spinal muscular atrophy type 3 (SMA type 3).
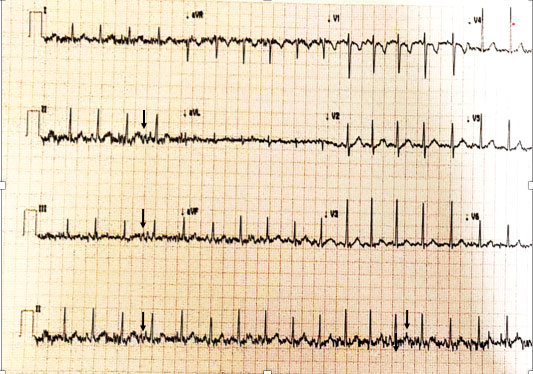 |
|
Fig. 1 Electrocardiogram of the
index patient showing the high frequency (30-40 Hz) tremor
(arrows) due to muscle fasciculations (seen predominantly in
limb leads).
|
Important differential diagnosis for progressive limb
girdle weakness presenting in late childhood (with onset beyond infancy)
include muscular dystrophies (especially Duchenne muscular dystrophy and
limb girdle muscular dystrophies) and SMA type 3. It may be difficult to
differentiate these conditions based on deep tendon jerks and creatine
kinase levels because these are often misleading. Deep tendon reflexes
may be preserved in SMA type 3 [1]. Joint hypermobility and hyperlaxity,
although an overlooked feature of SMA, if present favors a diagnosis of
SMA over muscular dystrophy [2,3]. The caveats include early-onset
muscle disorders such as congenital muscular dystrophies and congenital
myopathies [2]. In SMA, anterior horn cell loss begins in early infancy
and may possibly account for distal hypotonia and hyperlaxity.
Hyperlaxity, especially of upper limb joints may persist till adulthood
in more than half of patients [4]. It is perplexing to see that this
finding was not captured in major prospective cohorts of SMA type 2 and
3, which predominantly addressed the weakness and ambulation. This
finding needs to be further confirmed in large cohorts not only because
of diagnostic significance but also for rehabilitation point of view,
considering the improved outcomes with newer therapies in SMA.
REFERENCES
1. Lannaccone ST, Browne RH, Samaha FJ, Buncher
CR. Prospective study of spinal muscular atrophy before age 6 years.
Pediatr Neurol. 1993;9: 187-93.
2. Donkervoort S, Bonnemann CG, Loeys B, Jungbluth H,
Voermans NC. The neuromuscular differential diagnosis of joint
hypermobility. Am J Med Genet C Semin Med Genet. 2015;169C:23-42.
3. Haaker G, Fujak A. Proximal spinal muscular
atrophy: Current orthopedic perspective. Appl Clin Genet. 2013;6:113-20.
4. Tofts LJ, Elliott EJ, Munns C, Pacey V,
Sillence DO. The differential diagnosis of children with joint
hypermobility: A review of the literature. Pediatr Rheumatol Online J.
2009;7:1.
|
|
|
 |
|
