|
|
|
Indian Pediatr 2019;56:825-830 |
 |
Predictors of
Malnutrition in Children with Cystic Fibrosis
|
|
Nitin Dhochak, Kana Ram Jat, Jhuma Sankar, Rakesh Lodha and Sushil K
Kabra
From Department of Pediatrics, All India Institute of Medical
Sciences, New Delhi, India.
Correspondence to: Dr Sushil K Kabra, Department of Pediatrics, All
India Institute of Medical Sciences, Ansari Nagar, New Delhi 110029,
India.
Email: [email protected]
Received: March 27, 2019;
Initial Review: April 27, 2019;
Accepted: July 14, 2019.
Published online: August 10, 2019.
PII:S097475591600132
|
Objective: To determine occurrence of
malnutrition in children with cystic fibrosis and identify predictors of
malnutrition at time of enrolment and after 2 years of follow up.
Design: Retrospective chart review.
Setting: Pediatric chest clinic at a
tertiary-care center in northern India.
Patients: Cystic fibrosis patients enrolled
between 2009-2015 with at least 3 years follow-up.
Procedure: Weight and height were noted at
enrolment, and after 1 year and 2 years of follow-up. Clinical details,
medications, and pulmonary exacerbations during second year were
recorded.
Main outcome measure: Occurrence of malnutrition
i.e. weight for age Z-score < -2.
Results: 61 medical records were reviewed.
Occurrence of malnutrition at baseline, and 1- and 2-year follow-up was
65.5%, 54.1% and 57.3%, respectively. Weight for age Z-score at
enrolment significantly correlated with time to diagnosis from onset
r=0.015, P=0.029). Weight for age Z-score at 2-year follow-up was
significantly associated with steatorrhea (P=0.03), increased
frequency of stools (P<0.01) and pulmonary exacerbation (P=0.03)
during second year. Linear regression showed significant association
between weight for age Z-score at 2 years with steatorrhea and pulmonary
exacerbations [r=-0.795 (-1.527, -0.062)] and [r=-0.261 (-0.493,
-0.028)]. Pulmonary exacerbations during second and third year had
significant correlation with weight for age Z-score at the beginning of
respective years (r = -0.219, P=0.015).
Conclusion: Occurrence of malnutrition is high in
children with cystic fibrosis in this region, with uncontrolled fat
malabsorption and recurrent respiratory infections being significant
risk factors.
Keywords: Fat malabsorption, Nutrition, Pulmonary exacerbation.
|
|
C
hildren with cystic fibrosis (CF) are at
increased risk of malnutrition due to impaired absorption of nutrients
due to pancreatic insufficiency, increased basal energy requirement, and
recurrent pulmonary exacerbations, affecting growth. Other
co-morbidities like gastro-esophageal reflux disease, CF-related liver
disease, and CF-related diabetes can also contribute to inadequate
nutrition. Nutritional status is assessed with common anthropometric
parameters including weight, height and body mass index (BMI), and
calculating Z score from local growth standards [1]. In developed
countries, patients with CF have good age-appropriate nutrition with
good nutritional intake, pancreatic enzyme replacement therapy (PERT)
and high fat diet, but resource-poor countries persist to have
significant undernutrition [2-4]. Nutritional status has been associated
with respiratory health of the patients and nutritional interventions
like high fat/ high calorie diet have been shown to be temporally
related with improved nutrition and lung functions in studies from
United States and Canada [5].
In India, children are managed with basic
cost-effective therapies. Managing nutrition among children with CF in
such conditions is further challenging due to delayed diagnosis,
inadequate availability of specialist nutritional counselling, poverty,
and non-affordability of high cost PERT causing poor control of fat
malabsorption, and frequent infections [6]. This pre-disposes to higher
incidence of undernutrition, as seen in other developing countries [4].
There is paucity of studies on the extent of malnutrition and specific
predisposing factors operating in Indian subcontinent where CF is still
a relatively uncommon and emerging disease. The primary objective of the
study was to estimate occurrence of malnutrition in children with cystic
fibrosis. The secondary objectives were to determine predictors of
malnutrition at the time of enrolment in the clinic, and to determine
predictors of malnutrition after completing two years of follow up in
the clinic.
Methods
The study was approved by Institute Ethics Committee
of our institute with waiver of need for individual consent. It was a
retrospective chart review where records of all children with CF
enrolled in the Pediatric Chest Clinic of a tertiary-care centre in
Northern India between 2009 and 2015 were screened. Patients with at
least three years regular follow-up (at least two visits per year) after
enrolment, with regular weight and clinical records, were included in
the study. Cystic fibrosis was diagnosed based on suggestive clinical
history and two abnormal sweat chloride assays. Patients were advised a
high calorie and high fat diet. Children were regularly followed up in
the clinic at intervals of 1 to 3 months (depending on the clinical
condition). At each visit, anthropometric measurements, history (details
of respiratory and gastrointestinal symptoms) and examination, review of
medications and chest physiotherapy, and nutritional counselling were
done. Titration of PERT dose for fat malabsorption was done on the basis
of level of gastrointestinal symptom control (steatorrhea and diarrhea).
Anthropometric details (weight, and height or length)
recorded at enrolment, and at 1 year and 2 year of follow up were noted.
Z scores were calculated using age and sex appropriate growth standards
using "WHO Anthroplus" software. As weight was the most regularly
recorded parameter, weight for age Z score (WAZ) was taken as primary
parameter for assessing nutritional status. Patients with WAZ less than
–2 were classified as malnourished. Height/ length for age Z score (HAZ)
and body mass index Z score (BAZ) were also calculated.
Demographic details, age of onset of symptoms, time
taken to achieve diagnosis, sweat chloride value, and reports of the CF
gene mutation analysis were recorded for all patients. To identify
factors which contributed to poor nutrition in patients despite regular
CF-specific therapy, we studied parameters during second year of follow
up to identify predictors of nutrition at 2 year. These included
gastrointestinal symptoms, details of medications (lipase dose,
azithromycin, proton pump inhibitors, multivitamin supplements,
nebulisation drugs, and inhaled antibiotics), Shwachman-Kulczycki score
[7] and co-morbidities like gastroesophageal reflux disease, CF-related
diabetes, CF-related liver disease, intestinal obstruction, and allergic
bronchopulmonary aspergillosis (ABPA).
Pulmonary exacerbations were clinically diagnosed
based on increase cough, change of nature and amount of sputum, fever,
worsening dyspnea, and poor appetite; and treated with either oral or
intravenous antibiotics for at least 2 weeks depending on severity of
symptoms. Gastrointestinal symptoms (steatorrhea, diarrhea,
constipation, inadequate appetite) were considered significant if
present on at least two visits in the year. Pseudomonas colonization was
considered if two sputum cultures were positive for Pseudomonas
aeruginosa.
Statistical analyses: WAZ, HAZ and BAZ at
enrolment, 1 year and 2 year were compared using Wilcoxon sign rank
test. Association between WAZ and predictors were assessed using
Spearman’s correlation for continuous variables and Wilcoxon rank-sum
test for categorical variables, followed by linear regression. Overall
correlations between pulmonary exacerbations during second and third
year of follow-up with BAZ at the beginning of year were assessed using
Spearman’s correlation co-efficient. Analysis was done using STATA
software (StataCorp, College Station, TX).
Results
Sixty-one children (64% boys) were included in the
current study. Most of the children received regular chest physiotherapy
(98.3%), 3% saline nebulization (96.7%), PERT (98.3%), azithromycin
(96.7%) and fat-soluble vitamin supplementations (96.7%). None of the
children received DNase therapy. Median (IQR) number of visits during
second year were 4 (3, 5). Baseline characteristics of the cohort are
depicted in Table I. Clinical features during second year
of follow up including gastrointestinal symptoms and morbidities are
described in Table II.
TABLE I Clinical Features of Children with Cystic Fibrosis at Enrolment to Chest Clinic (N=61)
|
Clinical features |
No. (%) |
|
Male |
39 (63.9) |
|
#Age (mo) |
|
|
At enrolment |
22 (7, 60) |
|
At onset of symptoms |
2.7 (1, 6) |
|
#Time to diagnosis, mo |
19 (5.5, 48) |
|
*Mutation |
|
|
Homozygous del508 |
11 (33.3) |
|
Heterozygous del508 |
11 (33.3) |
|
Heterozygous 3849+10kb C>T |
1 (3.0) |
|
Negative for above |
10 (30.3) |
|
$Sweat chloride, mEq/L |
103 (27.6) |
|
Meconium ileus |
3 (4.9) |
|
Residence outside Delhi |
33 (54.1) |
|
*Done in only 33 patients; #median (IQR); $mean
(SD). |
TABLE II Clinical Features During Second-year Follow-up of Children with Cystic Fibrosis (N=61)
|
Clinical features |
No. (%) |
|
Gastrointestinal symptoms |
|
Steatorrhea |
20 (32.8) |
|
Constipation |
14 (22.9) |
|
Poor appetite |
14 (22.9) |
|
Diarrhea |
8 (13.1) |
|
Gastrointestinal co-morbidity |
|
GERD |
13 (21.3) |
|
CFLD |
9 (14.7) |
|
DIOS |
4 (6.6) |
|
Micronutrient deficiency |
2 (3.3) |
|
Respiratory morbidity |
|
Chronic colonization |
27 (44.3) |
|
Pseudomonas spp. |
26 (42.6) |
|
Staphylococcus spp. |
1 (1.6) |
|
ABPA |
4 (6.6) |
|
Pulmonary artery hypertension |
1 (1.6) |
|
Therapies |
|
$PERT (lipase units per kg per day) |
5684 (4039, 6694) |
|
Inhaled tobramycin |
14 (22.9)1 |
|
Inhaled colistin |
1 (1.6) |
|
Proton pump inhibitors |
14 (22.9) |
|
Ursodeoxycholate |
8 (13.1) |
|
Second year follow-up |
|
$Pulmonary exacerbation |
1 (0, 2) |
|
Patients hospitalized |
9 (14.8) |
|
Shwachman-Kulczycki score |
80 (75, 90) |
|
Third year follow-up |
|
$Pulmonary exacerbation |
1 (0, 2) |
|
Patients hospitalized |
12 (19.7) |
|
ABPA: allergic bronchopulmonary aspergillosis, CFLD: cystic
fibrosis related liver disease, DIOS: distal intestinal
obstruction syndrome, GERD: gastroesophageal reflux disease,
PERT: pancreatic enzyme replacement therapy. |
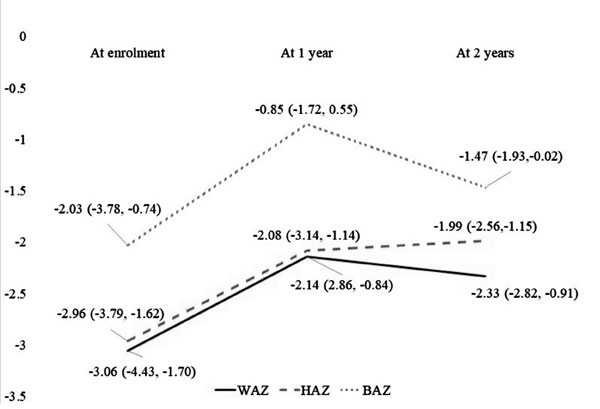 |
|
Fig. 1 Trends of anthropometric
parameters. Abbreviations: WAZ: weight for age Z score, HAZ:
height for age Z score, BAZ : body mass index for age Z score.
|
WAZ, HAZ and BAZ at baseline, and at 1 and 2 year
follow up are described in Fig. 1. WAZ (P<0.01),
HAZ (P=0.02) and BAZ (P=0.005) over first year showed
significant improvement. But during the second year, while improvement
was not seen in WAZ (P=0.89), HAZ showed improvement (P=0.02)
and BAZ showed decline (P=0.02). Occurrence of malnutrition (WAZ
<-2) at baseline, and at 1 and 2 year follow up was 65.5%, 54.1% and
57.3%, respectively.
Association of WAZ at enrolment with age of onset of
symptoms (r=0.24, P=0.07), time to diagnosis (r =0.16,
P<0.001), and sweat chloride (r = -0.09, P=0.52) was
variable. Baseline WAZ was also not associated with gender or place of
residence. Linear regression between WAZ with onset of symptoms, time to
diagnosis, gender and state revealed only time to diagnosis as
significant association (r=0.015, P=0.029).
Correlations between WAZ at 2 years of follow-up with
risk factors during second year i.e. numbers of pulmonary exacerbations
(r=-0.28, P=0.03), PERT (lipase intake per kilogram body weight)
(r=-0.25, P=0.05), number of oral antibiotic courses (r=-0.31,
P=0.02), number of IV antibiotic courses (r=0.19, P=0.14),
Shwachman-Kulczycki score (r=0.19, P=0.17), and number of
follow-up visits (r=-0.22, P=0.09), showed varying results.
Association between WAZ at 2 year and morbidities and therapies during
second year i.e. pseudomonas colonization, proton pump inhibitors
use, ursodeoxycholate use, steatorrhea, diarrhea, constipation,
appetite, Gastroesophageal reflux disease, CF liver disease, distal
intestinal obstruction syndrome, inhaled antibiotics, and pulmonary
arterial hypertension had P-value of 0.412, 0.686, 0.615, 0.031,
0.004, 0.162, 0.571, 0.744, 0.618, 0.704, 0.177 and 0.954,
respectively. Since steatorrhea and number of exacerbations were related
to diarrhea and number of oral antibiotic therapy, respectively, the
former were included in regression analysis. Linear regression between
WAZ at 2 years and risk factors i.e. pulmonary exacerbations,
steatorrhea, PERT dose and inhaled antibiotics showed significant
association with pulmonary exacerbations and steatorrhea (Table
III).
TABLE III Linear Regression of WAZ at 2 Years with Risk Factors
|
Variable |
Coefficient |
95% |
P-value |
|
|
Confidence |
|
|
|
interval |
|
|
Pulmonary exacerbations in second year |
-0.261 |
-0.493, -0.028 |
0.029 |
|
Steatorrhea |
-0.795 |
-1.527, -0.062 |
0.034 |
|
PERT dose (103 units lipase per kg per day) |
-0.100 |
-0.261, 0.061 |
0.216 |
|
Inhaled antibiotics |
0.606 |
-0.189, 1.402 |
0.133 |
|
Constant |
-0.904 |
-1.829, 0.041 |
0.060 |
|
R2=0.22; level of significance, P-value <0.05. PERT:
pancreatic enzyme replacement therapy, WAZ: weight for age Z
score. |
Overall pulmonary exacerbations during second and
third year showed significant association with WAZ (r = –0.22, P=0.01)
and BAZ (r= –0.26, P=0.006) at the beginning of second and third
year of follow-up; however, similar association was not found with HAZ (r=0.07,
P=0.48). While studying risk factors of pulmonary exacerbation of
second year, only WAZ at beginning of second year (P=0.03),
Shwachman-Kulczycki score (P=0.02), and pseudomonas colonization
(P=0.08) showed significance or trend to significance. Regression
analysis of these factors with pulmonary exacerbation during second year
demonstrated significant association with only Shwachman-Kulczycki score
(P=0.02) (Table IV).
TABLE IV Linear Regression of Pulmonary Exacerbations in Second Year with Predictors
Among Children with Cystic Fibrosis
|
Variable |
Coefficient |
95% |
P-value |
|
|
confidence |
|
|
|
interval |
|
|
WAZ at 1 year |
-0.197 |
-0.427, 0.032 |
0.090 |
|
Pseudomonas colonization |
0.647 |
-0.038, 1.333 |
0.064 |
|
Shwachman-Kulczycki score |
-0.037 |
-0.069, -0.006 |
0.020 |
|
Constant |
3.60 |
0.858, 6.344 |
0.011 |
|
RV2 = 0.22; level of significance P value <0.05;
WAZ: weight for age Z score. |
Discussion
From this evaluation of hospital records at a
referral center, we found that nearly two-thirds of newly enrolled
cystic fibrosis patients in our pediatric chest clinic are malnourished.
The nutritional status improved over initial one year, probably
secondary to the accelerated catch-up growth due to multiple
interventions integral to the care of CF children such as chest
physiotherapy, PERT, and fat-soluble vitamin supplementation. But the
nutritional status did not change much during the second year. This
implies that significant proportion of patients stayed malnourished even
after starting protocolized CF therapy with good compliance to daily
therapies.
Our results are different from that reported from the
developed countries. A study from Australia found mean WAZ score -0.14
in 1998 to up to 0.03 in 2014; while another multicentric study from
Europe had malnutrition in only 1.9% children [2,3]. Early diagnosis and
institution of multidisciplinary care in children diagnosed by newborn
screening of CF is associated with improved HAZ and WAZ than clinically
diagnosed children [8,9].
Scant data are available from developing countries.
Of two studies from Brazil, one showed median WAZ ranging from -0.18 in
infants to -1.26 in adolescents with progressive worsening with age,
while another study reported median BAZ at -0.58. The
Shwachman-Kulczycki score in these two studies was also better than our
cohort [4,10]. In another study form South Africa, the cohort had only
16% children below the third centile of weight for height [11].
Nutritional status of our cohort is poorer than reports from these
developing countries. Overall 35.8% under-5 years children in India have
WAZ<-2 as per 2015-2016 survey, which is significantly less than our
cohort of children with CF [12].
Nutritional status at enrolment to the chest clinic
showed weak positive correlation with time to diagnosis from onset of
symptoms. This seems paradoxical as delayed diagnosis should have had
worse nutritional [9]. Probably, the patients with delayed diagnosis in
our cohort, had less severe disease and hence less malnutrition.
Factors operating during second year of follow-up are
likely to impact the growth behavior during second year and hence,
nutritional status at 2 years. In our cohort, persistence of fat
malabsorption symptoms (steatorrhea and increased stool frequency) and
pulmonary exacer-bations were independently associated with worse WAZ at
2 years. High cost of PERT makes it difficult to achieve tight control
of fat-malabsorption. Lung functions and nutrition are inter-related and
follow similar trends; the poorly nourished patients are predisposed to
infections, and lung infections negatively impact growth, thereby
creating a vicious cycle [13]. Since most of our patients are
malnourished and have pulmonary morbidity at the time of enrolment to
clinic, it is difficult to establish cause-effect relationship.
Aggressive treatment of exacerbations and colonization along with
efforts to improve nutritional support should be done to break the cycle
of exacerbations and poor nutrition. Apart from the above studied risk
factors, lack of specialist CF nutritionist and underlying prevalent
malnutrition in our country could contribute to high incidence of
malnutrition in our cohort.
Worsening of nutritional status at the beginning of
the year is associated with pulmonary exacerbations in the following
year in our cohort. Aggressive nutritional intervention for one year in
children has been shown to decrease incidence of pulmonary exacerbations
[14]. A recent large study demonstrated significantly improved pulmonary
function with change in nutritional strategies form restricted fat to
high calorie–high fat nutrition [5]. Behavioral therapies, gastrostomy
feeding and parenteral nutrition are the different modes that have been
utilized to improve nutritional status but improvement in pulmonary
functions was demonstrated in the study with parenteral nutrition only
[15-17]. There is significant scope for aggressive dietary interventions
to improve nutritional and pulmonary health in our cohort.
Strength of our study is that the study addresses
population from Indian subcontinent where CF is still an emerging
condition and patients are managed with inadequate resources; also CF
patients in Indian subcontinent are genetically heterogenous from the
patients worldwide [18]. Limitation of our study include retrospective
study design leading to lack of detailed data on dietary intakes,
non-availability of fat malabsorption quantification, and limited
availability of pulmonary function test data.
We conclude that malnutrition is common in children
with CF in this region, which persists despite protocolized CF care.
Inadequate control of fat malabsorption is a significant predictor of
poor nutritional health. Recurrent respiratory infections during follow
up have significant negative impact on nutritional status. Apart from
overall awareness of CF diagnosis and treatment, interventions focused
on improving availability and affordability of PERT and reducing
infections, along with adequate nutritional support are likely to
improve nutritional status in CF children in Indian subcontinent.
Contributors: ND: contributed with conception of
work, acquisition, analysis and interpretation of data, drafting the
work; KRJ, JS, RL, SKK: contributed with conception of work,
interpretation of data, revising the work. All authors have approved the
final version and agreed to accountability for accuracy of work.
Funding: None; Competing interests:
None stated.
|
What is Already Known?
• Improvement in nutritional status is
associated with improvement in lung functions in children with
cystic fibrosis.
What This Study Adds?
• Children with cystic fibrosis in India are
at high risk of malnutrition.
• Uncontrolled fat malabsorption is an important predictor of
malnutrition in cystic fibrosis.
|
References
1. Turck D, Braegger CP, Colombo C, Declercq D,
Morton A, Pancheva R, et al. ESPEN-ESPGHAN-ECFS Guidelines on
Nutrition Care for Infants, Children, and Adults with Cystic Fibrosis.
Clin Nutr. 2016;35:557-77.
2. Calvo-Lerma J, Hulst JM, Asseiceira I, Claes I,
Garriga M, Colombo C, et al. Nutritional status, nutrient intake
and use of enzyme supplements in paediatric patients with Cystic
Fibrosis; a European multicentre study with reference to current
guidelines. J Cyst Fibros. 2017;16:510-8.
3. Ruseckaite R, Pekin N, King S, Carr E, Ahern S,
Oldroyd J, et al. Evaluating the impact of 2006 Australasian
Clinical Practice Guidelines for Nutrition in Children with Cystic
Fibrosis in Australia. Respir Med. 2018;142:7-14.
4. Hauschild DB, Barbosa E, Moreira EAM, Ludwig Neto
N, Platt VB, Piacentini Filho E, et al. Nutrition status
parameters and hydration status by bioelectrical impedance vector
analysis were associated with lung function impairment in children and
adolescents with cystic fibrosis. Nutr Clin Pract. 2016;31:378-86.
5. Goss CH, Sykes J, Stanojevic S, Marshall B, Petren
K, Ostrenga J, et al. Comparison of nutrition and lung function
outcomes in patients with cystic fibrosis living in Canada and the
United States. Am J Respir Crit Care Med. 2018;197:768-75.
6. Kabra SK, Kabra M, Lodha R, Shastri S. Cystic
fibrosis in India. Pediatr Pulmonol. 2007;42:1087-94.
7. Shwachman H, Kulczycki LL. Long-term study of one
hundred five patients with cystic fibrosis; studies made over a five- to
fourteen-year period. AMA J Dis Child. 1958;96:6-15.
8. Sims EJ, Clark A, McCormick J, Mehta G, Connett G,
Mehta A, et al. Cystic fibrosis diagnosed after 2 months of age
leads to worse outcomes and requires more therapy. Pediatrics.
2007;119:19-28.
9. Siret D, Bretaudeau G, Branger B, Dabadie A,
Dagorne M, David V, et al. Comparing the clinical evolution of
cystic fibrosis screened neonatally to that of cystic fibrosis diagnosed
from clinical symptoms: a 10-year retrospective study in a French region
(Brittany). Pediatr Pulmonol. 2003;35:342-9.
10. Neri L de CL, Bergamaschi DP, Silva Filho LVRF da.
Evaluation of nutritional status in patients with cystic fibrosis
according to age group. Rev Paul Pediatr. 2019;31:58-64.
11. Westwood AT, Saitowitz R. Growth and nutrition in
South African children with cystic fibrosis. S Afr Med J.
1999;89:1276-8.
12. National Family Health Surveys (NFHS) in India:
Lessons learnt and way forward. Available from:http://rchiips.org/NFHS/pdf/NFHS4/India.pdf.
Accessed November 28, 2018.
13. Steinkamp G, Wiedemann B. Relationship between
nutritional status and lung function in cystic fibrosis: Cross sectional
and longitudinal analyses from the German CF quality assurance (CFQA)
project. Thorax. 2002;57: 596-601.
14. Shepherd RW, Holt TL, Thomas BJ, Kay L, Isles A,
Francis PJ, et al. Nutritional rehabilitation in cystic fibrosis:
Controlled studies of effects on nutritional growth retardation, body
protein turnover, and course of pulmonary disease. J Pediatr.
1986;109:788-94.
15. Watson H, Bilton D, Truby H. A randomized
controlled trial of a new behavioral home-based nutrition education
program, "Eat Well with CF," in adults with cystic fibrosis. J Am Diet
Assoc. 2008;108:847-52.
16. Efrati O, Mei-Zahav M, Rivlin J, Kerem E, Blau H,
Barak A, et al. Long term nutritional rehabilitation by
gastrostomy in Israeli patients with cystic fibrosis: clinical outcome
in advanced pulmonary disease. J Pediatr Gastroenterol Nutr.
2006;42:222-8.
17. Allen ED, Mick AB, Nicol J, McCoy KS. Prolonged
parenteral nutrition for cystic fibrosis patients. Nutr Clin Pract.
1995;10:73-9.
18. Shastri SS, Kabra M, Kabra SK, Pandey RM, Menon PSN.
Characterisation of mutations and genotype-phenotype correlation in
cystic fibrosis: Experience from India. J Cyst Fibros. 2008;7:110-5.
|
|
|
 |
|
