|
|
|
Indian Pediatr 2016;53: 917-919 |
 |
Cerebrotendinous
Xanthomatosis Without Skin Changes: Diagnostic Delay and
Confirmation by Genetic Analysis
|
|
Shilpa D Kulkarni, Meenal Garg and Rafat Sayed
From Department of Pediatric Neurosciences, Bai
Jerbai Wadia Hospital for Children, Mumbai, India.
Correspondence to: Dr Shilpa Kulkarni, EEG room, 2nd
floor, Department of Pediatric Neurosciences, Bai Jerbai Wadia Hospital
for Children, Parel, Mumbai 400 012, Maharashtra, India.
Email:
[email protected]
Received: December 30, 2015;
Initial review: March 07, 2016;
Accepted: May 19, 2016.
|
Background: Cerebrotendinous xanthomatosis is an
inherited lipid storage disease manifesting with infantile onset
diarrhea, cataracts, xanthomas and adult-onset neurological dysfunction
with cerebellar signs and neuropathy. Case: 10-year-old boy
presented with progressive ataxia, neuropathy and cataracts. Over 6
years, he developed dementia, kyphoscoliosis with worsening ataxia, and
neuropathy. Outcome: Sterol analysis and CYP27A1
sequencing confirmed the diagnosis.Message: The condition should
be considered in childhood onset cerebellar ataxia with cataracts, even
in the absence of skin signs.
Keywords: Childhood-onset ataxia; Lipid metabolism, Neuropathy.
|
|
Cerebrotendinous xanthomatosis (CTX; OMIM#213700)
is a lipid storage disease characterized by infantile-onset diarrhea,
childhood-onset cataract, tendon xanthomas and adult-onset progressive
neurologic dysfunction [1]. It is an autosomal recessive disorder caused
by mutations in the CYP27A1 gene located on chromosome 2q33,
leading to reduced production of chenodeoxycholicacid (CDCA) and
increase in cholestanol levels [2,3]. We describe a child who presented
with early onset neurological symptoms without xanthomas.
Case Report
A 10-year-old boy presented with progressive gait
imbalance. He was the third child born of a third-degree consanguineous
marriage and had two normal elder siblings. There was no family history
of neurological disease. He had an uneventful birth. Mother gave history
of diarrhea in the first month of life which settled after oral
medication. Developmental milestones were delayed in all four domains.
He had poor scholastic performance with writing difficulties. He also
had a refractive error since 6 years of age and wore spectacles. There
was no history of seizures. He had progressive difficulty in climbing
stairs and gait imbalance since 8 years of age. He had recently started
having falls while walking. On examination, he was able to understand
and follow simple instructions. His height was 120 cm (less than 3 rd
percentile) and head circumference was 51 cm (between 10th
and 25th percentile). There
were no skin lesions. He was noted to have bilateral developmental
cataracts. Speech was slurred but understandable, and cranial nerve
examination was normal. There was high stepping gait and bilateral pes
cavus. Deep tendon reflexes were depressed, power was 4/5 in proximal
muscles and 3/5 in distal muscles. Cerebellar signs were predominant
including ataxia, dysmetria, intention tremors and dysdiadokinesia.
Romberg’s sign was positive.
Patient was first investigated to rule out treatable
causes of chronic progressive ataxia (Vitamin E levels, peripheral
smear, fasting lipid profile, lactate levels, etc). Lipid profile was
normal with cholesterol of 170 mg/dl (normal levels-122-228 mg/dL).
Differential diagnoses of cataracts with progressive ataxia were
considered at this point, including Friedreich ataxia, Marinesco-Sjogren
syndrome, CTX and Congenital Cataracts with Facial Dysmorphism and
Neuropathy. Echocardiogram was normal. Magnetic Resonance Imaging (MRI)
of the brain showed bilateral parieto-occipital white matter
abnormalities with dentate nucleus hyperintensities (Fig.1).
Nerve conduction studies suggested symmetrical sensorimotor
demyelinating polyneuropathy. He was treated symptomatically without any
benefit. Sequencing of FXN gene for Friedreich ataxia did not
reveal any pathogenic variation. Further biochemical and genetic testing
could not be carried out owing to financial constraints.
 |
|
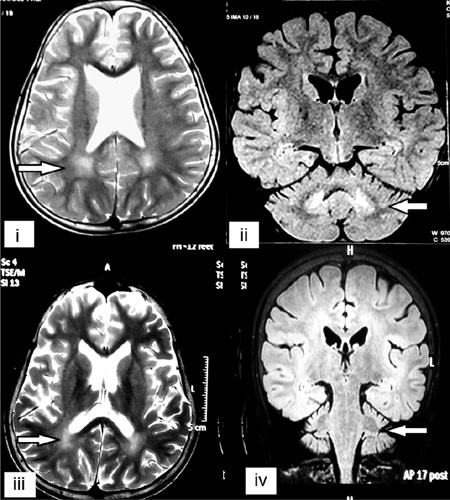
Fig. 1 (i). T2-weighted axial section
of MRI brain showing parieto-occipital white matter hyperintense
signal. (ii). Fluid attenuated inversion recovery (FLAIR)
coronal section showing dentate nucleus involvement and
cerebellar atrophy. (iii). T2-weighted axial section of MRI
brain done 4 years later showing increase in parieto-occipital
white matter hyperintense signal and cortical atrophy (iv).
FLAIR coronal section showing increase in dentate nucleus
involvement and cerebellar atrophy.
|
The patient was followed up for six years. Ataxia and
slurring of speech slowly worsened. Cataracts were operated. He
developed dementia and dropped out of school. There was appearance of
kyphoscoliosis along with distal wasting, increasing pes cavus and new
onset dystonia. X-rays did not show any significant abnormality
except spinal deformity. Dual energy X-ray absorptio-metry scan
showed a Z score of -4.0 (lower than normal). Pulmonary function tests
were normal. MRI of the brain repeated after four years showed worsening
of the changes in dentate nucleus and white matter with cerebellar
atrophy (Fig. 1). Sterol analysis revealed high
cholestanol (69.98 umol/L, normal value: 0.04-9.31 umol/L), normal
cholesterol (1,916.97 umol/L; normal value: 1050-3229 umol/L) and
cholestanol:cholesterol ratio of 0.03651 (normal: ~0.0051). Sequencing
of the CYP27A1 gene was performed and showed c.525delG homozygous
mutation resulting in a frameshift and premature truncation of the
protein. This is a previously reported mutation in a patient with CTX
[4] and was confirmatory of the clinical suspicion. The parents were
advised genetic testing but were unwilling in view of financial
concerns. Genetic testing for siblings has been advised.
Discussion
Patients with CTX lack the mitochondrial enzyme
sterol 27-hydroxylase leading to reduced levels of CDCA and up
regulation of the enzyme precursors of bile acid formation pathway and
cholestanol. Neurological involvement is seen in adolescents and adults
in the form of cerebellar and supratentrorial symptoms, myelopathy,
epilepsy, parkinsonism, psychiatric manifestations and peripheral
neuropathy. Cardiopulmonary disease and osteoporosis are other features
[6].
Our patient presented with early onset ataxia,
cataracts and neuropathy. Treatable causes such as abetalipoproteinemia,
vitamin B 12 and vitamin E
deficiencies were ruled out. Friedreich ataxia was considered as it is
one of the most common causes of recessively inherited ataxia and has
myriad presentations. Marinesco-Sjogren syndrome can also present with
early onset cataracts, cerebellar signs, cerebellar atrophy and
psychomotor retardation, but patients have early onset myopathy.
Congenital contract with facial dysmophism and neuropathy similarly
presents with cataracts, neuropathy, late onset cerebellar signs, short
stature and mild facial dysmophism; our patient did not have dysmorphic
features or significant anterior chamber abnormalities.
Early onset of cerebellar signs with polyneuropathy
without xanthomas has been rarely described in CTX. Due to the absence
of xanthomas, there was a delay in the diagnosis. A strong possibility
was reconsidered after application of clinical suspicion index [7], but
could not be confirmed at the time.
CTX should be considered as a differential diagnosis
of chronic progressive ataxia in pediatric age group. Cataracts and
early refractory diarrhea may act as important clues. Xanthomas may
develop later in the course. A multidisciplinary treatment approach and
use of CDCA and statins is recommended. Genetic counseling and testing
for asymptomatic members should be offered to the family.
Contributors: SDK, MG: data acquisition;
SDK, MG: literature research; SDK, MG, RS: manuscript preparation; SDK,
MG, RS: manuscript editing and revision. All authors have read the
manuscript and approved the final version.
Funding: None; Competing Interest: None
stated.
References
1. Federico A, Dotti MT, Gallus GN. Cerebrotendinous
Xanthomatosis. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE,
Amemiya A, Bean LJH, et al., editors. GeneReviews [Internet].
Seattle (WA): University of Washington, Seattle; 1993-2015.
2. Cali JJ, Hsieh CL, Francke U, Russell DW.
Mutations in the bile acid biosynthetic enzyme sterol 27-hydroxylase
underlie cerebrotendinous xanthomatosis. J Biol Chem. 1991;266:7779-83.
3. Fraidakis MJ. Psychiatric manifestations in
cerebrotendinous xanthomatosis. Transl Psychiatry. 2013;3:302.
4. Verrips A, Steenbergen-Spanjers GC, Luyten JA, van
den Heuvel LP, Keyser A, Gabreëls FJ, et al. Two new mutations in
the sterol 27-hydroxylase gene in two families lead to cerebrotendinous
xanthomatosis. Hum Genet. 1996;98:735-7.
5. Pilo-de-la-Fuente B, Jimenez-Escrig A, Lorenzo JR,
Pardo J, Arias M, Ares-Luque A, et al. Cerebrotendinous
xanthomatosis in Spain: clinical, prognostic, and genetic survey. Eur J
Neurol. 2011;18:1203-11.
6. Nie S, Chen G, Cao X, Zhang Y. Cerebrotendinous
xanthomatosis: a comprehensive review of pathogenesis, clinical
manifestations, diagnosis, and management. Orphanet J Rare Dis.
2014;9:179.
7. Mignarri A, Gallus GN, Dotti MT, Federico A. A
suspicion index for early diagnosis and treatment of cerebrotendinous
xanthomatosis. J Inherit Metab Dis. 2014;37:421-9.
|
|
|
 |
|
