e describe the clinical and autopsy findings of a
7-month-old baby with complaints of repeated respiratory infections and
failure to thrive.
Clinical Protocol
History: A seven-month-old boy presented with
complaints of fever and cough for four months and tachypnea for 1 week
prior to admission. He was born to a third gravida by normal vaginal
delivery with birthweight of 3 kg and an uneventful perinatal period. He
was immunized for age. He had been symptomatic from four months of life
and was hospitalized thrice for pneumonia and treated with intravenous
antimicrobials prior to admission in PGI. He was not thriving well, with
history of oral ulcers for past four months. One unit of un-irradiated
blood was transfused 10 days prior to admission in PGI.
Family history revealed death of his father at 45
years of age due to electrocution. He had two elder male siblings who
were alive and well. There was a history of death of two maternal uncles
at age 6 and 11 months due to repeated infections. There was no history
of contact with pulmonary tuberculosis.
Clinical examination: His weight was 3.5 kg (<-3Z
score); length; 63 cms (at -3Z score); and head circumference: 38 cms
(<-3Z score). He was afebrile with a pulse rate of 142/min which was
regular and good volume. Respiratory rate was 68/min; blood pressure;
80/50 mm Hg; capillary filling time, 2 sec; and SaO2 on pulse oximetry :
76%. He looked emaciated with severe pallor and dry skin. There was no
icterus, clubbing or rash. BCG scar was absent. Oral cavity examination
revealed thrush.
Both lung fields revealed bilateral crepitations. No
organomegaly or lymphadenopathy was detected. The cardiovascular system
examination was unremarkable. On central nervous system examination, he
was noted to be irritable but there were no focal neurological deficit,
or signs of meningeal irritation.
Provisional diagnosis: Failure to thrive, and
Repeated chest infections. A possibility of cystic fibrosis (CF) or
immunodeficiency was considered.
Laboratory Investigations: His hematological,
biochemical, coagulation and blood gas analyses during hospital stay are
detailed in Tables I and II.
Microbiology work up showed:
Blood cultures (Day 2 and Day 4): sterile
Fungal blood culture (Day 3): sterile
Endotracheal (ET) tube aspirate (Day 3):
Klebsiella pneumoniae
Gastric lavage for acid-fast bacilli (AFB) (Day 2 and
Day 3): negative
Mantoux test: non-reactive
Urine: routine examination was normal; Urine fungal
smear: negative
HIV ELISA: Non-reactive; Cytomegalovirus (CMV) IgM:
positive
Primary immunodeficiency work up: Immunoglobulin
profile (sent after blood transfusion): IgM < 11 mg/dL (normal 40-160
mg/dL); IgG 786 mg/dL (normal 300-900 mg/dL); IgA 61 mg/dL (normal 15-70
mg/dL).
Lymphocyte subset analysis by flowcytometry: CD3
- 45.7%; CD19 - 1.6%; CD16 - 21.7%. Impression: Both T and B lymphocyte
numbers are significantly decreased; consistent with T-B-NK+ SCID
(severe combined immunodeficiency).
TABLE I Blood and Coagulation Parameters During the Stay
|
Dates
|
Hb (g/dL) |
TLC |
DLC (%) |
Absolute lymphocyte
|
Others |
|
|
(per cmm) |
P/L/M/E |
count (per mm3) |
|
|
1 month before presentation* |
8.5 |
10,600 |
– |
– |
Plat Ct. 1,67,800/mm3 |
|
15 day before presentation* |
7.8 |
11,200 |
62/28/6/4 |
3136 |
– |
|
Day 3* |
5.7 |
7,000 |
23/67/7/3 |
4690 |
– |
|
Day 1* |
8.4 |
7,900 |
32/58/7/3 |
4582 |
– |
|
Day 1 |
7 |
5,300 |
– |
–
|
Plate Ct. 73,000/mm3 |
|
Day 2 |
7.8 |
3,200 |
82/3/13/2 |
96 |
Plat Ct. 81,000 /mm3 |
|
Day 3 |
7.2 |
4,300 |
76/3/20/1 |
129 |
Plat Ct. 1,32,000/mm3 |
|
Day 4 |
|
|
|
|
PT 18s; PTI 72%;
|
|
|
|
|
|
APTT 31s; d-dimer
|
|
|
|
|
|
positive |
|
Day 5 |
6.3 |
5,200 |
90/3/6/1 |
156 |
Plat Ct. 74,000/mm3 |
*hemograms done outside before admission in our hospital;
PT-Prothrombin time;
APTT: activated partial thromboplastin timb;
PTI: prothrombin index; P- polymorphs; L- lymphocytes;
M- monocyles; E- eosinophils. |
TABLE II Biochemical Parameters of the Patient
|
Parameters
|
Day 1 |
Day 2
|
Day 4 |
Day 6 |
|
Na/K (mEq/L) |
123/3.7 |
146/3.5 |
145/4.6 |
146/5.5 |
|
Urea/creatinine (mg/dL) |
30/0.2 |
23/0.3 |
76/0.6 |
166/1.1 |
|
S. protein (g/dL); A/G |
- |
5.1; 2.4/2.7 |
5.1; 2.6/3.5 |
- |
|
S. bilirubin (mg/dL) |
- |
0.7 |
0.7 |
- |
|
CRP (mg/L) |
- |
32 |
47 |
- |
|
pH |
7.33 |
- |
7.4 |
7.05* |
|
PaO2 (mmHg) |
102 |
- |
63 |
55* |
|
PaCO2 (mmHg) |
44 |
- |
40 |
49* |
|
HCO3 (mmol/L) |
23 |
- |
25 |
13* |
|
S: Serum; A: albumin; G: globulin; *on day 7. |
Radiology: The initial X-rays done outside
showed hyperinflated lungs with definite opacities in parahilar
location. Subsequently, there was an increase in the homogenous
opacities. Based on the X-rays, presence or absence and size of
thymus were difficult to comment upon but it was definitely not
enlarged. Subsequent X-rays done in our hospital showed
consolidation with air bronchogram in bilateral lung fields. There was
evidence of right middle lobe collapse and consolidation. Repeat X-rays
showed evidence of pneumomediastinum. Another X-ray, done 2 days
before death, showed bilateral diffuse homogenous opacification of both
lung fields with extensive air bronchograms.
Course in hospital: The baby presented with
respiratory failure for which he was intubated and put on manual IPPR.
He was maintained on manual IPPR throughout the hospital stay. Though
his initial SaO2 was maintained around 92%, from day three onward of
hospital stay it was persistently below 80%. He was started on broad
spectrum antimicrobials (ceftriaxone, amikacin, cloxacillin), which were
subsequently changed to imipenem (ET aspirate report – Klebsiella
spp). In view of the possibility of fungal infection (as evident from
oral thrush) amphotericin was also started. He was transfused with
packed red cell 10 mL/kg to correct anemia. With worsening sepsis and
hypoxemia, he developed shock on day five of hospital stay that was
managed with fluid boluses and inotropes. However, the shock was
refractory and he developed oliguric renal failure and suffered a
cardiac arrest from which he could not be revived.
Unit’s final diagnosis: Persistent pneumonia
with sepsis, oral candidiasis. A possibility of cystic fibrosis and
primary immunodeficiency–SCID is considered.
Discussion on clinical protocol: We have a
baby who is not thriving well since 3 months of age. So, the illness
starts well in the first 6 months of age. He had repeated episodes of
pneumonia with oral thrush. In fact, none of the chest X-rays
done outside were normal. So, with this combination of symptoms
immunodeficiency is very likely. Virtually there is no differential
diagnosis for an infant who presents with this combination of symptoms.
If there is immunodeficiency, this can be primary or secondary. HIV
infection, which is the commonest cause of secondary immunodeficiency
disease, was ruled out by the treating unit. There were no other obvious
causes of secondary immunodeficiency like a malignancy or exposure to
glucocorticoids. So, most likely what we are dealing with is primary
immunodeficiency (PID) in this child which has started in the first few
months of life.
How do you suspect PID?
The Jeffrey Modell foundation has suggested 10
warning signs of a PID and this is the most well accepted guideline
available to clinicians to suspect an immunodeficiency (Box I).
|
Box I Warning Signs for PID |
1. Four or more new ear infections within 1 year.
2. Two or more serious sinus infections within 1
year.
3. Two or more months on antibiotics with little
effect.
4. Two or more episodes of pneumonia within 1 year.
5. Failure to gain weight or grow normally.
6. Recurrent deep skin or organ abscesses.
7. Persistent oral thrush or fungal infection on
skin.
8. Need for intravenous antibiotics to clear
infections.
9. Two or more deep-seated infections including
septicemia.
10. A family history of PID. |
If a child has two or more of these signs then PID is
more likely. In our patient six of these signs were present. So it is
more than likely that this child had PID manifesting in the first 6
months of life.
What is the likely type of PID here?
We have a 7-month-old baby with failure to thrive and
repeated hospitalizations for pneumonia requiring several courses of
intravenous antimicrobials. These clinical manifestations would be
common to any immunodeficiency presenting in infancy, but the giveaway
is the fact that he became symptomatic at 3-4 months of age and children
who become symptomatic so early in life usually have a cell-mediated
immuno-deficiency. SCID is the syndrome of cell-mediated
immunodeficiency which presents so early.
In addition, there is a family history of an X-linked
recessive disorder- two maternal uncles dying in infancy with repeated
infections. This is consistent with a type of SCID, the X-linked SCID –
which affects only boys and the mother is usually the carrier.
On physical examination, the baby had severely
impaired anthropometric parameters because of the illnesses he had
suffered for last several months. There was no lymphadenopathy. Absence
of lymphadenopathy can occur in 3 types of PID – X-linked
agammaglobulinemia, SCID and hyper IgM syndrome. There is perhaps no
other condition where you can get absent lymph nodes in a baby. We
cannot comment on the tonsils. But again if tonsils were also absent, it
is these three conditions where tonsillar tissue is not seen. He had
oral thrush, which is indicative of a cell-mediated immunodeficiency,
i.e. SCID.
There are significant pointers in the history and
physical examination supporting the diagnosis of PID. On investigations
there was significant lymphopenia. The lower limit of normal lymphocyte
count from birth to 6 months is 2000/cmm; 12 months it is 4000/cmm. An
absolute lymphocyte count as low as 2000/cmm during infancy is highly
suggestive of SCID. Our patient had decreased T cell numbers. The serum
immunoglobulin showed an IgM which was undetectable and lymphocyte
subset analysis showed T-B-NK+ phenotype. So overall the investigations
are supportive of a diagnosis of SCID. However, we must realise the
limitations to these available investigations. We have not performed a
lymphocyte proliferation response to mitogens, like phytohemaglutinin,
which would have confirmed the diagnosis of SCID as lymphocytes in SCID
do not proliferate to mitogen response. Similarly, we do not have an
antibody response after immunization because the baby was so sick this
could not be done, and most importantly, mutation analysis is not
available.
SCID is one of the most difficult of all PIDs to
diagnose clinically and is perhaps the only immunodeficiency where
definition incorporates mutation analysis as part of the enumeration.
Here, we only have clinical features and laboratory investigations
consistent with the diagnosis of X-linked SCID.
What other PIDs need to be excluded?
Agammaglobulinemia is unlikely as the baby presented
very early. Children with agammaglobulinemia become symptomatic after 6
months of life. In hyper IgM-syndrome, the IgM levels should be
elevated. Here the IgM was absent, so this is unlikely. For chronic
granulomatous disease (CGD) we do not have the reports of NBT
dye-reduction or the DHR-shift on flowcytometry. These children have
leucocytosis and hypergammaglobulinemia and the clinical picture is very
different. Wiskott-Aldrich syndrome (WAS) could be considered because
this was a male and had persistent thrombocytopenia. However, in the
absence of microthrombocytes in the peripheral smear and WAS protein in
flowcytometry, this could be a clinical possibility, though less likely.
SCID is a syndrome and not a disease. There are at
least 13 different types of SCID and it is impossible to differentiate
one from the other without mutation analysis.
What are the common infections that are expected in
SCID?
You could have many bacterial infections of both gram
positive and gram negative organisms. This child had received BCG, so
disseminated BCG infection can occur in this baby. Pneumocystis
jirovecii infection can again be very prominent feature of SCID.
Among the viral infections (this baby had received at least 2
un-irradiated blood transfusions), CMV could be expected as CMV IgM was
positive.
The final diagnosis would be SCID with superadded
infections- disseminated BCG, CMV, fungal and bacterial infections. But
I must say that, the morphological correlates of SCID on autopsy are
very few. SCID is not a diagnosis which you make on autopsy. The only
morphological correlate would be an absent thymus or a thymus which is
below 1g and this thymus would show an absence of Hassall’s corpuscles
and absent corticomedullary differentiation. There may be absent lymph
nodes and absence of lymphocytes in Peyer’s patches.
Dr. Meenu Singh: Whenever a patient with recurrent or
persistent pneumonia like this comes to our unit we give slightly wider
range of diagnosis to be ruled out especially after the nutritional and
the chronic infection parts are taken care off. Cystic fibrosis (CF) is
unlikley because of the presence of persistent pneumonia and
hyperinflation in this particular child. As the investigations have
revealed, it is prudent to think of PID at the outset and with the
history of two maternal uncles’ death in infancy, possibility of SCID
was thought of and investigations were carried out. As far as the
superadded infections are concerned, disseminated BCG infection is a
common finding in children with SCID. However, in the beginning, one
should certainly rule out CF by doing sweat chloride test. This was
probably not followed in this child as the main working diagnosis was an
immunodeficiency.
Dr. Sunit C Singhi: I am not going to dispute the
diagnosis that has been forwarded. I think in patient who comes at 4
months with episodes of repeated infections, the diagnosis of CF is not
out especially with the chest X-rays showing hyperinflation. The
first and second chest X-rays showing a shadow of the shape of
thymus. But if it has completely involuted, one does not know now. On
the top of that, we have counts from outside which showed lymphocytes to
be normal which are difficult to explain. Is it because of the
overwhelming infections that had exhausted the lymphoid tissue? It could
be disseminated tuberculosis which could do all of it and it could be
even any overwhelming infection. I think normal lymphocyte counts needs
an explanation and to begin with at least there should be no reason not
to consider other differential diagnosis causing recurrent pneumonias
and CF
Dr. Sanjay Jain: With the simultaneous presence of
hepatitis, pneumonitis, pancytopenia with diarrhea, diagnosis of
disseminated cytomegalovirus infection (CMV) is straight-forward.
Dr. Joseph L Mathew: in our unit we arranged a date
for sweat chloride test. Because he was on ventilator, we could not get
it done. Secondly, the ten criteria put forth by Jeffrey Modell
foundation to suspect PID, is a very sensitive definition. In other
words, all children with PID will have a score of >2. Then it loses
specificity in the sense that, there are infants with two episodes of
pneumonia in a year who get treated with two rounds of antibiotics, they
too land up in getting screened for PID with that criteria. The third
point is on one side the infant is not producing IgM because his total
IgM is only 11mg/dl. So, to say that he had CMV infection and he
produced CMV IgM positivity would be very hard. Perhaps it is the blood
transfusion which is related to this.
Dr. Varun Dhir: The strong family history just points
to an X-linked disorder and with the lymphocyte count being so low the
diagnosis virtually stands as SCID. Only query is why the
immunophenotype demonstrating T-B-NK+ type. In fact, with absolute
lymphocyte count of 150 or 100/cmm, any percentage of NK cells come as a
T-B-NK. This would lead to a diagnosis of ADA deficiency type of SCID.
But that is not consistent with the family history of the patient. In
X-linked SCID there would be a common gamma chain. One way, we could
have done it by doing common gamma chain expression by flowcytometry,
which is available.
Dr. Surjit Singh: The lymphocyte subsets in a patient
with SCID is very difficult to interpret. Here, there was absolute
lymphocytopenia. So, in this setting, even though some lymphocytes
stained for CD3, the number was so low that we interpreted this as
T-B-NK+. This is not the phenotype of X-linked SCID. X-linked SCID is
T-B+NK-. I really cannot explain why this baby did not show some B
cells. But SCID is a very difficult condition to enumerate without
mutation analysis. If the lymphocyte counts are as low as these,
possibly there is no differential diagnosis. However, in a patient with
SCID, the lymphocyte count can go up under certain situations and the
most common situation is, when there is maternal engraftment of T-cells.
As this was a male, if there were maternal engraftment of T-cells, the
lymphocyte counts can be high. There are ways to differentiate whether
these lymphocytes are the baby’s lymphocytes or they belong to the
mother. Normally, a SCID child does not mount an antibody response
provided it is an absolute SCID, which happens in ADA deficiency
(T-B-NK). These children usually would not have an antibody response to
any infection. Here CMV IgM was positive. We do not know the titre
whether-low titres or high titres. Several SCIDs have leaky syndrome
that means they have some lymphocytes which can manifest with either an
antibody response or some immunoglobulin. The immunoglobulins in this
patient were never zero. Some studies have shown that some children with
SCID have relatively higher immunoglobulins.
Pathology Protocol
A partiaA partial autopsy was performed on this 7-month-old
male child. The peritoneal and pleural cavities revealed presence of
ascites and pleural effusion with 250ml of straw-colored and 150ml of
hemorrhagic fluid, respectively.
The thymus weighed 2 g (normal for this age-12 g),
and was small and atrophic (Fig. 1A). The lobular
configuration was lost and cortico-medullary distinction was not
discernible on histology. The epithelial cells were present in diffuse
sheets with scant Hassall’s corpuscles (Fig. 1B). There
was marked depletion of lymphocytes with focal areas showing abundance
of mast cells (Fig. 1C). All these features are indicative
of thymic dysplasia which is noted in patients of SCID.
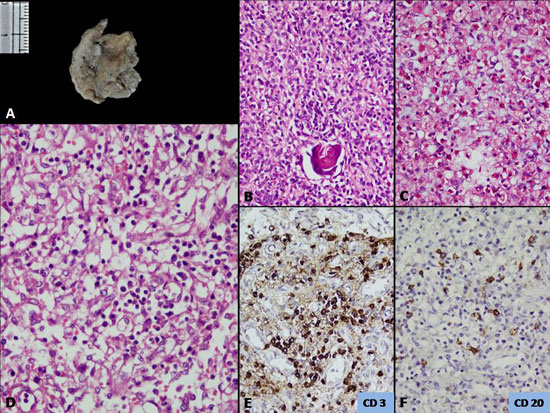 |
|
Fig. 1 (a) Small and atrophic thymus
with loss of lobular configuration, (b) Epithelial cells
in diffuse sheets and scant Hassall’s population and depleted
lymphoid cells (H&E x400); (c) Increased number of mast
cell within the thymic parenchyma (H&E x400); (d) Lymph
node with absent of lymphoid follicles and germinal centre and
marked depletion of lymphocytes (H&E x400); and (e & f) CD3 and
CD20 immunostaining reveals scant population of T- and B-cells,
respectively (immunoperoxidase ×400).
|
All the lymphoid rests of the body, including the
lymph nodes, revealed depletion of lymphoid population. A small lymph
node identified in the peri-pancreatic region showed almost complete
absence of lymphoid follicles and germinal centre with prominence of
sinuses (Fig.1D). The T cell zones i.e. the paracortical
areas revealed scant population of T lymphocytes which were highlighted
by immunoreactivity with CD3 (Fig. 1E). CD20
immunoreactivity highlighted the markedly depleted B lymphocytes (Fig.
1F). The s). The spleen weighed 18 g (normal for this age-25 g). On
histology, there was capillarization of sinuses. The peri-arteriolar
sheath, which are the T cell zones, were depleted. The submucosal
lymphoid aggregates in appendix and Peyer’s patches were represented by
small clusters. The Peyer’s patches were hard to identify on gross
examination. Lymphoid depletion was also identified in the section taken
from the bone marrow.
The lungs were heavy weighing 180 g (normal for
age-110 g). The mucosa over the tracheo-bronchial tree was congested;
all the airways were filled with inspissated secretions. Both lungs
revealed diffuse consolidation with a firm texture and loss of
crepitancy of lung parenchyma (Fig. 2A). No areas of
breaking-down abscesses or caseous necrotic foci were detected. On
histology, most of the alveolar spaces were packed with granular
eosinophilic secretions which were periodic acid-Schiff (PAS) positive.
In other areas, instead of the granular material, the alveolar spaces
were packed with foamy macrophages with finely vacuolated cytoplasm
which were positive with Oil Red ‘O’ (Fig. 2B). The
features are indicative of endogenous lipoid pneumonia developing
secondary to obstruction. Interstitial fibrosis and inflammation was
minimal. At places, the inspissated secretions had tracked down to the
respiratory bronchioles and alveolar spaces with surrounding parenchyma
showing features of bronchopneumonia and collections of Gram negative
cocci. In addition, many multinucleated giant cells were identified
within the alveolar spaces (Fig. 2C). Occasional CMV
inclusions were noted within the alveolar lining cells (Fig.
2D). Squamous metaplasia within the bronchiolar lining was noted at
places. Furthermore, features of diffuse alveolar damage with formation
of hyaline membrane were identified. Polymerase chain reaction (PCR)
done for detection of Respiratory Syncytial Virus (RSV) was performed.
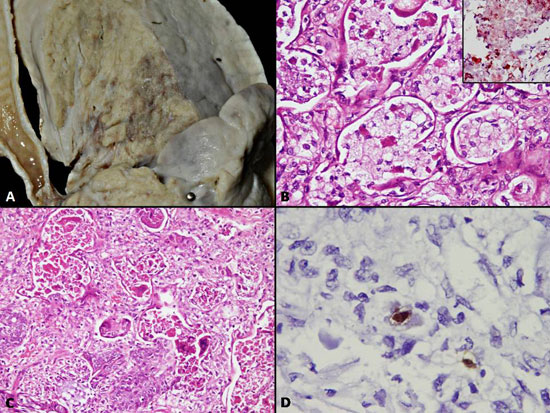 |
|
Fig. 2 (a) Gross pictures of cut
surface of right lung showing diffuse consolidation; (b)
Alveolar spaces packed with finely vacuolated macrophages (H&E
x400); inset – Positivity with oil-Red-‘O’ (H&E x400); (c)
Multinucleated giant cells lining the alveolar spaces (H&E
x400); and (d) Occasional Cytomegalovirus inclusion detected
with immunostain for CMV antigen. (immunoperoxidase ×400).
|
In the lung, no granulomas were identified and
Ziehl-Neelsen stain for AFB was negative. The liver weighed 158gms
(normal for this age-280gms) and did not reveal any significant
pathology. No features to suggest cystic fibrosis were identified in
liver and pancreas microscopically. Similarly, vas deferens and testes
were normal. Large and small intestine did not reveal any inspissated
secretions. Kidneys were fused at the lower pole with isthmus present
posterior to the aorta. Adrenals weighed 1.5 g (normal for this age-5.5
g), on microscopic examination revealed cytomegalovirus inclusions
within the adrenal cortical cells. Heart including other organs was
within normal limits on gross and microscopic examination.
Dr Mini P Singh: The lung tissue which we received
was subjected to RNA extraction using the ethod, and subjected
it to RT-PCR for both the human RSV and human metapneumovirus (MPV). The
sample was positive for RSV with a band corresponding to 680 base pair
length of DNA and negative for MPV.
Autopsy diagnosis
A case of severe combined immunodeficiency with
• Thymic dysplasia with marked lymphoid depletion
in lymph nodes, spleen, appendix, Peyer’s patches, and bone marrow
• Bronchopneumonia and diffuse alveolar damage
(due to RSV)
• Endogenous lipoid pneumonia
• Cytomegalovirus (CMV) inclusion in lungs and
adrenals
• Horse-shoe kidney
• Ascites and pleural effusion
Open Forum
Dr Sunit C Singhi: I have several queries. Regarding
the diagnosis as it was put forward was not in doubt. But, it does not
explain how the BCG did not cause a disease here, which could not happen
with SCID as far as I know. The lymphocyte counts done outside in the
first 2 months were normal. Is it because they don’t do it or is there
any explanation for these normal counts? In this age group, the CMV
infection usually involves the lungs. If it is not there, there would be
an explanation for this.
Dr SK Jindal: How long after the BCG vaccination
there can be recrudescence? Normally, after BCG inoculation the bacilli
disappear after few weeks and subsequently the immune reaction which is
there and which is the purpose of BCG vaccination. In this case, the
vaccination was given at birth and the child developed infection after
several months. So, disseminated BCG infection could not be considered
as a clinical diagnosis.
Dr Meenu Singh: Regarding the endogenous lipoid
pneumonia as it was presented, it was following an obstruction. Is it
the mucous causing bronchiolar obstruction or is it because of RSV
infection which can lead to obstruction and lipoid pneumonia? Mostly
lipoid pneumonias are because of milk aspiration as milk contains fat
globules.
Dr Pratibha D Singhi: No significant infection other
than RSV and CMV were present. The presence of gross ascites with straw
colour fluid and pleural effusion are not usual features of RSV
infection alone. Do we have any explanation for this?
Dr Kirti Gupta: Detection of CMV inclusion in the
lungs was very difficult and these were picked up on
immunohistochemistry. Regarding lipoid pneumonias, it includes 2 types –
the endogenous and exogenous. The exogenous one is due to milk
aspiration whereas; the endogenous one occurs due to obstruction of the
major airways. So, there was excess production of mucous because of
infections and this mucous got reabsorbed and collected in macrophages.
And this is not CF as in CF the impaction of thick viscid secretions
occurs in the bronchial glands not in the major airways like in this
case.
Dr S Prabhakar: Is there any explanation for the
three reports of lymphocyte counts done outside which mentioned
lymphocyte counts of 28-58%?
Dr Surjit Singh: SCID is associated with low
lymphocyte counts and the more severe the immunodeficiency the lower
would be the lymphocyte counts. The absolute lymphocyte count in fact
can be used for the screening for SCID. Any baby in the first 12 months
of life, who has an absolute lymphocyte count below 3000 is very likely
to have SCID, and any baby who has less than 2000 has to have SCID.
Absolute lymphocyte counts are frequently missed as paediatricians
mostly focus on neutrophils. Newborn screening for SCID is now a
reality, because this is a true medical emergency and if transplantation
is done in these babies within a few months of life, chances of survival
are 80-90%. Why this baby did not have disseminated BCG infection even
when the BCG vaccine had been given? There could be many reasons for
this. SCID is not a disease, it is a syndrome and it depends on how
severe is the immune deficiency. There are all kinds of phenotypes which
can manifest as SCID. Depending on the T-cell response one may or may
not have infection with one or more of the organisms. So, it is not
surprising that BCG has not disseminated.
Dr RK Ratho: Regarding CMV IgM positivity, in
contrast to the cut-off value of 0.3 this patient had 1 OD. It is
definitely quite high. Patient with SCID with CMV IgM positivity, having
CMV inclusions imply that it was an active response to infection rather
than due to transfusions. Also, suppose the patient was diagnosed with
RSV infection during life, what would be the way of treatment? Do you
have ribavarin treatment or other prophylactic measures?
Dr Meenu Singh: Probably we have to educate the
public regarding immune deficiency. In presence of a suspicious family
history, one should definitely go for screening procedures. Common
infections encountered in immune deficient patients are RSV. It is more
severe in patients who have underlying lung disease, immune deficiency,
CF, heart disease etc. In these patients ribavarin is recommended.
Especially in patients who are to be put on ventilator, a special
circuit is connected for administering ribavarin. Our patient was on
ambu bag ventilation and not on a mechanical ventilator.
Dr S Prabhakar: Pediatricians see these cases more
frequently; for others, it is not such a common disease and this case
was instructive.
Discussion
Primary immunodeficiency disorders (PIDs) comprise
more than 150 different disorders that affect the development, function,
or both of the immune system [1]. All forms of PIDs are rare and have an
overall prevalence of approximately 1:10,000 live births with the
exception of IgA deficiency. PIDs are classified into eight major
categories (Box 2) according to the component of the
immune system primarily involved [1].
|
Box 2 Classification of Primary Immuno-deficiency
Disorders |
1. Combined T-cell and B-cell immunodeficiencies
2. Predominantly antibody deficiencies
3. Other well defined immunodeficiency syndromes
4. Diseases of immune dysregulation
5. Congenital defects of phagocyte number and function
6. Defects in innate immunity
7. Autoinflammatory disorders
8. Complement deficiencies |
SCID disorders involve both B- and T- cells and
includes about 22 different groups of diseases within its category
[1,2]. Most of the B-cell abnormalities appear secondary to the lack of
T-cell help, which underscores the role of T-cell in B-cell development.
Inheritance of this congenital syndrome may show X-linked or autosomal
recessive pattern. Affected infants are highly susceptible to recurring
infections of viruses, fungi and bacteria and invariably die within 2
years of birth. The patients usually present within first six months of
life with failure to thrive, chronic diarrhea, persistent oral thrush,
skin rash, pneumonia, and sepsis. Disseminated BCG infection is commonly
seen in patients with SCID. Similarly, prolonged interstitial pneumonia
of viral etiology such as parainfluenza virus or cytomegalovirus or
Pneumocystis jerovicii is also common in patients with combined
immunodeficiency [3,4].
The autopsy findings in the index case highlight the
morphological changes encountered in patients with SCID–thymic
dysplasia, hypoplasia of spleen, lymph nodes and marked depletion of
lymphocytes in all the lymphoid reservoirs of the body. Mast cell
hyperplasia within the thymus is an interesting morphologic feature
reported in SCID patients early in the literature [5]. The patients are
prone to repeated respiratory infections secondary to immunodeficiency.
RSV has been earlier reported in autopsy series in SCID infections [6].
It usually manifests with presence of multinucleated giant cells lining
the alveolar spaces. Infection with CMV in SCID has also been
well-documented in the literature [7]. Rare examples of endogenous
lipoid pneumonia, developing secondary to obstruction have been
documented in autopsy cases of SCID [8]. In endogenous lipoid pneumonia
fat-filled finely vacuolated macrophages fill the alveoli as seen in the
index case. CF was excluded based on absence of characteristic
morphologic findings at autopsy for instance- secondary biliary
cirrhosis, small atrophic gall bladder, inspissated secretions within
pancreatic ducts, meconium ileus and absence of vas deferens.
Endogenous lipoid pneumonia in autopsy series of SCID
patients has seldom been documented in the literature and the index case
in addition to the other features, adds observational data to the
examples of SCID autopsy patients reported in the literature.
References


