|
|
|
Indian Pediatr 2009;46: 897-899 |
 |
Pachyonychia Congenita With Unusual Features |
|
S Balasubramanian, K Kaarthigeyan and B Ramnath
Department of Pediatrics, Kanchi Kamakoti CHILDS Trust
Hospital, Chennai, India
Correspondence to: Dr S Balasubramanian, Senior Consultant Pediatrician, Kanchi Kamakoti CHILDS
Trust Hospital,
12-A, Nageswara road, Nungambakkam, Chennai 34, TN, India.
Email: sbsped53@sify.com
Manuscript received: June 18, 2008;
Initial Review: July 29, 2008;
Accepted: November 21, 2008.
|
|
Abstract
Pachyonychia congenita is a rare hereditary disorder
characterized by gross thickening of all finger and toenails. We report
an infant who had clinical features consistent with pachyonychia
congenita type II, with unusual features of microcephaly, seizures,
electroencephalogram abnormalities, failure to thrive, and heterochromia
iridis.
Key words: Congenital dyskeratosis, Nail dystrophy,
Pachyonychia congenital.
|
|
P
achyonychia
congenita (PC) is a rare hereditary disorder characterized by nail
dystrophy, palmar and plantar hyperkeratosis, leukokeratosis of the mucous
membranes, follicular keratosis, and occasional hyperhidrosis of palms and
soles(1). We report an infant with PC type II, who also had unusual
features.
Case Report
An 11-month old male infant, 1 st
born of 2nd degree consanguineous parentage presented with abnormal nails
and skin lesions since birth, along with delayed motor and mental
milestones and poor weight gain. There was history of seizures at 3rd
month of life. There was no similar nail and skin changes in any other
family member in two generations. There was no history of natal teeth.
Anthropometric measurements revealed grade II protein-energy malnutrition
(IAP classification) and microcephaly (head circumference 42 cm). The
infant had sparse and thin hair over the scalp, heterochromia of iris, low
set ears, angular cheilosis, with a hoarse voice and stridor. All the
fingernails and toenails were dystrophic, discolored, and thickened, with
massive subungual hyperkeratosis producing distal elevation of nail plates
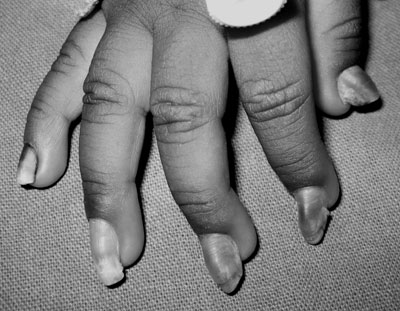
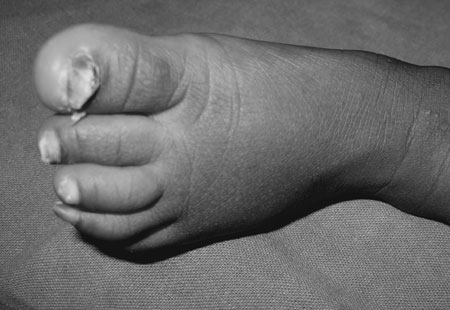
with wedge-shaped deformity of the nails (Fig. 1 and
2). He also had pin-head sized follicular papules over the elbows,
knees, popliteal and gluteal areas. Marked hyperhidrosis was observed in
the palms and soles. Oral cavity was normal with 4 primary teeth.
Neurological examination revealed spasticity of all the limbs, with
scissoring of lower limbs, dystonia and pseudo-bulbar palsy. A complete
hemogram, serum biochemistry and liver enzymes were normal. Urine
metabolic screening tests were negative. Karyotyping was normal.
Electroencephalogram was abnormal with evidence of seizure activity from
left hemisphere. Computed tomography (CT) of head revealed cerebral
atrophy. Direct laryngoscopy showed moderate degree of laryngomalacia with
mild congestion of vocal cords. He was treated with haloperidol, sodium
valproate, emollients and parents were counseled regarding rehabilitation.
 |
 |
|
Fig. 1 Nails grown to full length with an
upward slant caused by the predominant distal hyperkeratosis giving
‘wedge-shaped’ nails. |
Fig. 2
Thickened and dystrophic toenails. |
Discussion
Pachyonychia congenita has both autosomal dominant and
autosomal recessive forms of inheritance, reflecting heterogeneity(2). PC
encompasses a range of inherited ectodermal dysplasias, divided into two
main subtypes, Pachyonychia congenita type 1 (PC-1, Jadassohn-Lewandowski
syndrome) and Pachyonychia congenita type 2 (PC-2, Jackson-Lawler
syndrome), which can usually be distinguished by clinical examination.
Considerable overlap can occasionally exist between PC-1 and PC-2, which
can make diagnosis difficult. This most recent classification of PC is
based on clinical findings and by molecular genetic testing(3).
The predominant and most common clinical feature in PC
is hypertrophic nail dystrophy. Other findings common to PC-1 and PC-2
are: focal palmoplantar keratoderma, blistering, oral leuko-keratosis, and
palmoplantar hyperhidrosis, follicular keratosis on trunk and extremities,
and piloseba-ceous cysts. Other findings observed in PC-2 are
steatocystoma which normally develops at puberty, pili torti (hair
anomalies) and natal teeth(3). The pattern of Pachyonychia congenita
correlates well with the keratin gene mutation. The two keratin genes
known to be associated with PC-1 are KRT6A and KRT16 and the two keratin
genes known to be associated with PC-2 are KRT6B and KRT17(3). PC should
be differentiated from traumatic thickening of nails and from congenital
onychogryphosis, which are easy to recognize because they do not involve
all nails and are not associated with dyskeratotic skin lesions. Based on
the clinical manifestation, our cae appear to be type II, as per recent
classification(3). Hoarseness is a feature of PC-1 but may also be seen in
PC-2 along with unruly hair. The relationship of mental retardation to
Pachyonychia congenita is not clear. It has been suggested that the fetal
ectodermal lesions that affect the skin may also affect the central
nervous system(4). Our case in addition had other unusual features namely
microcephaly, seizures(1), electro-encephalogram abnormalities(5) and
failure to thrive(4), which have been described in few previous isolated
case reports. However, a combination of all these unusual features has not
been described earlier. The presence of heterochromia iridis may be
another new association of pachyonychia congenita or may just be a chance
occurrence.
Treatment of PC primarily provides symptomatic relief.
Emollients, keratolytics, topical retinoids and oral retinoic acid are
used to treat palmoplantar hyperkeratosis(6). The only effective treatment
for nail lesions is surgery with radical excision of the nail, nail bed,
and nail matrix, and skin implantation at the site of the removed nail(7).
Contributors: SBS, KK were involved in case
management. KK, BR were involved in review of literature and preparation
of manuscript. SBS will act as guarantor.
Funding: None.
Competing interests: None stated.
References
1. Feinstein A, Friedman J, Schewach-Millet M.
Pachyonychia congenita. J Am Acad Dermatol 1988; 19: 705-711.
2. Su WP, Chun SI, Hammond DE, Gordon H. Pachyonychia
congenita: a clinical study of 12 cases and review of the literature.
Pediatr Dermatol 1990; 7: 33-38.
3. Smith FJ, Kaspar RL, Schwartz ME, McLean WH,
Leachman SA. Pachyonychia Congenita. Gene Reviews. http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=pc.
Accesed 31 August, 2008.
4. Laing CR, Hayes JR, Scharf G. Pachyonychia congenita.
Am J Dis Child 1966; 111: 649-652.
5. Akesson HO. Pachyonychia congenita in six
generations. Hereditas 1967; 58: 103-110.
6. Rohold AE, Brandrup F. Pachyonychia congenita:
therapeutic and immunologic aspects. Pediatr Dermatol 1990; 7: 307-309.
7. Caproni M, Fabbri P. Pachyonychia congenita.
Orphanet Encyclopedia. http://www.orpha.net/data/patho/GB/uk-PachyonychiaCongenita.pdf.
Accessed 31 August, 2008.
|
|
|
 |
|
