|
|
Letters to the Editor Indian Pediatrics 2006; 43:923-924 |
|||
|
Oligomeganephronia in Wolf- Hirschhorn Syndrome |
|||
|
A two-year-old male was admitted with the complaints of facial dysmorphism, multiple congenital abnormality and proteinuria. The child was born of nonconsanguineous marriage and family members were healthy. On the physical examination; the weight (7 kg), the height (78 cm) and the head circumference (41 cm) were less than third percentile. The blood pressure was 70/45 mmHg. Increased antero-posterior skull diameter with bitemporal narrowing, medially thin eye-brows, hypertelorism, down-slanting palpebral fissure, hemangiomas on the right eyelid and neck, unilateral cataract, bilateral iris coloboma, beaked nose, high palat, deep philtrum and microretrognathia were determined. Laboratory evaluation revealed 2(+) proteinuria with dipstick. The urinary protein excretion was 200 mg/day (23 mg/m2/h) with a volume of 200 mL/day. Urea (24 mg/dL), creatinine (0.4 mg/dL), serum albumin (4.1 g/dL), cholestrol (197 mg/dL), and triglyceride levels (140 mg/dL) were in normal ranges. The visible chromosomal deletion at the 16.3 region of the short arm of the chromosome 4 [46, XY, del (4p16.3)] was detected with conventional cytogenetic methods in the patient, except the other members of the family (de novo deletion).
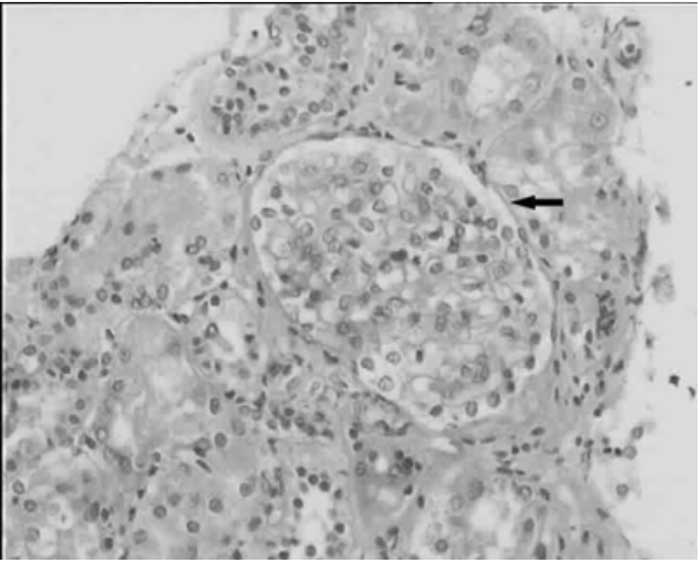
Kidneys were hyperechogeneic and small sized on abdominal ultrasonography. The kidney biopsy revealed one hypertrophic glomerulea (4-fold enlarged than others), of five (Fig.1). The tubules were hypertrophic. The amyloid, immunoglobulin, complement deposition, or interstitial infiltration were not detected. The reduction in proteinuria was facilitated by reninangiotensin system antagonism. The size and the site where anchored of the deleted part of the chromosome 4 reveal the phenotypic aspects in WHS. Characterization of the genes encoded in the WHS 4p break point region and role of these genes in the development may provide the insight necessary to understand the pathogeneic mechanisms of involvements in this syndrome(2). Oligomeganephronia is characterized by a severe developmental defect of both kidneys without urinary tract malformation. Typically, the size of the kidneys are small (20-75% of the normal), and the affected nephrons are 20- 25% of the total with hypertrophic tubules and thickening of Bowman’s capsule. To the best our knowledge, oligomeganephronia with 4p deletion syndrome (syndromic) have solely been reported in 5 fetuses and 2 cases with variable numbers of midline fusion defects at autopsy(1,3). The association of oligomega-nephronia with proteinuria is thought to be due at least partly to the hyperfiltration and increased glomerular size that raises the stress on the glomerular capillary wall(4). Because of their preferential action on the efferent arteriolar tone, renin-angiotensin system antagonists reduce intraglomerular pressure and proteinuria. Moreover, angiotensin antagonism suppresses local growth factor, cytokine and chemokine release, with subsequent reduction of glomerular hypertrophy and sclerosis as well as tubulointerstitial inflammation and fibrosis(5). Onur Sakallioglu,
|
![]()