Griscelli syndrome (GS), a rare autosomal recessive
disorder, results in pigmentary dilution of hair, immunodeficiency and
recurrent lymphohistiophagocytosis (LHP) (1,2). The terminal event is
usually an accelerated phase (LHP) similar to Chediak-Higashi
syndrome(CHS). However, the hallmarks of CHS, the giant cytoplasmic
granules in circulating granulocytes, are not found. We report a child
with GS highlighting differences between CHS and GS and the unusual
features of a Dandy-Walker cyst and hypergammaglobulinemia.
Case Report
A six-year-old girl presented with recurrent fever,
seizures and regression of milestones since 4 years. She also had
anasarca and jaundice since 3 months. A first born of a third-degree
consanguineous union, her 4-year-old male sibling was normal and there
was no family history of immuno-deficiency.
On examination she was chronically undernourished,
febrile, edematous, icteric and pale. She had a striking
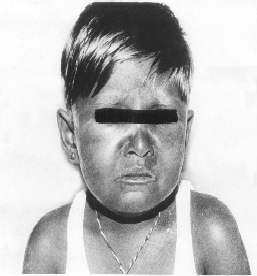
hypopigmentation (silvery-gray sheen) of her scalp hair, eyebrows and
eyelashes (Fig. 1). There was a horizontal nystagmus, normal iris
and retinal pigmentation. She had a massive spleno-hepatomegaly with no
free fluid. There was generalized hypertonia, hyperreflexia, lower limb
power was grade 3/5 compared to upper limbs (4/5) and no involuntary
movements. Her IQ was 67 and other systems were normal. In view of
recurrent fever, pallor, organo-megaly and silvery gray hair a
differential diagnosis of Chediak-Higashi versus Griscelli syndrome was
considered.
 |
|
Fig. 1. Clinical photograph shows silvery
grey sheen of eyebrows, eyelashes and scalp hair.
|
Investigations revealed pancytopenia with Hb 6.2 g/dL,
TLC 1000/cmm [N48%, L52%, (ANC of 480)], platelets 31,000/cmm and 0.4%
reticulocytes. The peripheral smear was normocytic hypochromic and
confirmed pancytopenia. The liver function revealed abnormal
hypoalbuminemia (2.1 g/dL), alka-line phosphatase (1196 mg/L), and gamma
GT (459 mg/dL). Blood culture was sterile. Mantoux was non-reactive and
urine examina-tion was normal. Hypertriglyceridemia (347 mg/dL) and
elevated immunoglobulins (IgG 1411, IgA 210, IgM 156 mg/dL) were
detected. The absolute lymphocyte subsets were low [CD4 186 (59%) and
CD8 107 (34%)]. Bone marrow was mildly hypo-cellular with erythroid
hyperplasia, moderate megaloblastic changes and depleted iron stores.
There was no evidence of lympho-histiocytic infiltration or giant
granules in any of the cell line series. HIV, HBsAg, HCV and Paul-Bunnel
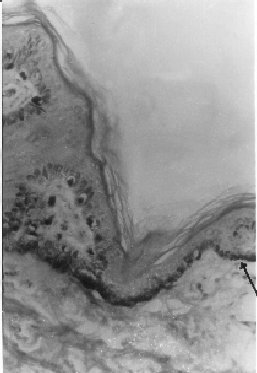
tests were negative. A CT scan showed a Dandy-Walker cyst. Skin biopsy (Fig.
2) revealed increased pigment in the basal skin layer containing
melanocytes with some clumping. There was poor pigmentation of adjacent
keratinocytes. Also seen was irregular unevenly distributed large
clumping of pigment primarily in the hair medulla.
 |
|
Fig. 2. Skin biopsy (HE stain × 100 oil)
shows increased pigment in the basal layer containing melanocytes
with some clumping. There is poor pigmentation of adjacent
keratinocytes. |
In view of the pigment dilution, recurrent
infections, neurological findings, typical skin and hair pathology and
the absence of coarse granules in the neutrophils a diagnosis of
Griscelli Syndrome was made. She was treated with blood components,
intravenous antibiotics, carbamazepine, iron, folate and vitamin B12 as
a bone marrow transplant was unaffordable. The child was lost to follow
up after the initial discharge.
Discussion
Griscelli syndrome is characterized by pigment
dilution, immunodeficiency, neuro-logical impairment and a virus
associated accelerated phase of lymphohistiocytosis with
hemophagocytosis. CHS has a similar clinical presentation but has giant
neutrophilic granules with minimal neurological involve-ment. A skin
biopsy and hair mount help to confirm the diagnosis. In CHS, a skin
biopsy would show the presence of giant melanosomes in the melanocytes
on electron microscopy and pigment dilution in both the melanocytes and
keratinocytes. Hair would differ by showing evenly distributed, small,
granular aggregates.
This child with GS had nystagmus and normal eye
pigmentation, which could indicate a cerebellar involvement. Her
neurological deterioration started abruptly at 2 years following an
acute febrile episode. Subsequently, there was gradual partial recovery
of truncal strength enabling her to sit up by the time she presented to
us. However, her lower limb weakness continued. The Dandy-Walker (DWC)
malformation pro-bably compounded her neurological impair-ment (cerebellar)
but the spasticity and seizures were due to GS in view of the small size
DWC. Neurological impairment is a known prominent feature in many
patients with GS (3-5). Clinical manifestations can include intracranial
hypertension(6), cere-bellar signs(6), encephalopathy, hemi-paresis,
peripheral facial palsy, spasticity(l), hypo-tonia(3,4), seizure(l),
psychomotor retarda-tion(3-5), and progressive neurological
deterioration(1,4,5). The pathology involves cerebral lymphohistiocytic
infiltration and erythrophagocytosis with nonspecific EEG patterns.
Neuroradiological studies have shown increased signal intensity
involving both gray and white matter(1), and white matter changes
primarily in the posterior fossa(3). This association of GS and DWC has
never been previously published in literature.
The frequent infections in this child were probably
due to abnormal T cell function and neutropenia. The unusual feature was
associated hypergammaglobulinemia. The common finding is a normal or a
hypo-gammaglobulinemia(2). One possible expla-nation for this unusual
finding could be that our child had repeated infections causing a
polyclonal hypergammaglobulinemia, assum-ing she had a normal
gammaglobulin level to begin with. Hepatitis C liver disease with
associated hypergammaglobulinemia was also ruled out. Granulocyte
abnormalities in GS are sporadic and inconsistent, with normal
intracellular oxidative metabolism(6). Giant cytoplasmic granules in
leucocytes are consistently absent.
The accelerated phase is commonly preceded by an
infection with the Ebstein-Barr virus, but can also be triggered by
infection with Hepatitis A virus or bacteria(6). This is characterized
by fever, hepatospleno-megaly, pancytopenia, hemophagocytosis,
coagulo-pathy, elevated serum transaminase levels, hypoproteinemia and
elevated triglycer-ides(6). Our patient had fever, pancytopenia,
hepatosplenomegaly, coagulopathy and hypertriglyceridemia with negative
viral screens. However, there was no evidence of lymphohistiocytosis in
the bone marrow and a liver/spleen biopsy could not be done in view of
the severe coagulopathy. Treatment involves the use of steroids,
etoposide, cyclosporine, antithymocyte globulins and intrathecal
methotrexate(7,8) in various combinations but with poor results.
Though two genes have been mapped to chromosome
15q21, the antenatal diagnosis of GS is only possible by morphological
criteria of examining fetal scalp hair(9). The prognosis for patients
with GS is grave. Bone marrow transplant is the only hope for a cure if
performed early in the course of the disease before the development of
accelerated phase(10). In view of the poor prognosis, there is a role
for genetic counselling and attempts at prenatal diagnosis in both GS
and CHS.
Contributions: CD and SL clinically diagnosed,
drafted and revised the report. KRK and SRH assisted in confirming and
documenting the pathological diagnosis.
Funding: None.
Competing interests: None stated.
