|
|
Brief Reports Indian Pediatrics 2001; 38: 1148-1154 |
||||
|
Glutaric Aciduria Type I |
||||
|
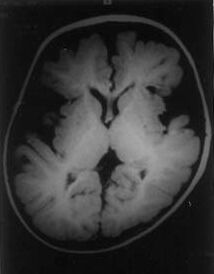
Glutaric aciduria Type I (GA-I) is a recessively inherited inborn error of metabolism characterized by the deficiency of the mitochondrial enzyme Glutaryl CoA dehydrogenase (GCDH) which catalyzes the dehydrogenation - decarboxylation of Glutaric acid, an intermediary metabolite in the degradation pathway of lysine, hydroxy-lysine and tryptophan. Excessive accumula-tion of Glutaric acid and other metabolites is supposed to be responsible for the manifesta-tions in this neurodegenerative metabolic disease(1). This communication describes the clinical profile of four patients with this condition. Case Reports Case I This 18-month-old boy was born of a third degree consanguineous marriage to a sixth gravida mother after an uneventful pregnancy and labor. Developmental milestones (social smile and head control) were normal till 3 months of age when he suffered an acute viral illness characterized by loose motions, fever and irritability. This was followed by a loss of already achieved milestones and recurrent generalized tonic-clonic seiures. The child stopped recognizing the mother and gradually developed spasticity of the limbs. A previous sib had died at 3 months of age, presumably of gastroenteritis. There was failure to thrive (weight and height below the 5th percentile) and microcephaly (head circumference 44 cm, less than 5th percentile). The neuro-logical examination revealed left-sided hemiparesis, spasticity, dystonic posturing and hyperactive deep tendon reflexes. Myocolonic seizures were also observed. The CT Scan of the brain showed enlarged peri-Rolandic CSF spaces. There were ill-defined hypodense areas in the basal ganglia. The MRI of the Brain confirmed these findings (Fig.1) and also revealed extensive white matter abnormalities in the cerebral hemi-spheres. The plasma and urinary amino-acidograms were normal. Urine analysis by gas chromatography-mass spectroscopy (GC-MS) revealed a marked excretion of glutaric acid and trace excretion of 3-hydroxyglutaric acid. The diagnosis of GA-I was confirmed on the basis of characteristic neuroimaging and biochemical studies. Therapeutic intervention in the form of a low protein diet with restriction of lysine and tryptophan along with administration of high doses of riboflavin and L-carnitine was offered. Valproate was used for seizure control. When last seen on follow-up at 24 months of age, the child’s weight had increased by 800 g. The child was seizure free and the dystonia had reduced. He had regained social smile, head control and was attempting to roll over.
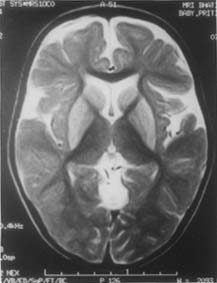
Case 2 The propositus was a 2-year-old girl born following an uncomplicated pregnancy to parents who were first cousins. Her development was normal till 2 years of age when she experienced sudden loss of head control, intellectual deterioration and onset of generalized tonic-clonic seizures following a mild febrile illness with gastroenteritis. She had microcephaly (head circumference 45.5 cm, below 5th percentile) and the neurolgoical examination disclosed limb hypotonia, complete absence of head control and choreiform movements affecting the distal portions of the extremities. The MRI of the brain revealed areas of hypointensity within the putamen and caudate nucleus on T1-weighted imagesx. On the corresponding T2 weighted (Fig. 2), proton and FLAIR sequences these areas appeared hyper-intense. The peri-Rolandic CSF spaces were enlarged and large CSF spaces were noted anterior to the temporal lobes. These abnormalities were highly suggestive of GA-I. The arterial blood gas analysis showed mild metabolic acidosis with an increased anion gap. Ketone bodies were absent in the urine. There was no hypoglycemia or hyper-ammonemia. GC-MS examination of urine detected an impressive excretion of glutaric acid and a smaller peak of 3-hydroxyglutaric acid, confirming the diagnosis of GA-I. urine and plasma aminoacidograms were normal. She was advised dietary treatment along with riboflavin, carnitine and phenytoin. A year later the girl was sitting up independently, was able to feed herself and obeyed simple commands. The dystonia had decreased although intermittent opisthotonus was noted and hypertonia perisisted.
Case 3 This was the 4-year-old elder male sibling of Case 2. Following an unremarkable pregnancy and postnatal period, the parents noticed developmental delay and abnormal posturing of limbs and tonic seizures at 7 months of age. Phenobarbitone had been pres-cribed for seizure control. When examined by us at 4 years of age, there was failure to thrive and microcephaly (head circumference 47.5 cm, below the 5th percentile). Head holding and approach to objects were absent. On neurological examination there was dystonia, orofacial dyskinesia and athetoid movements of the limbs with generalized spasticity. The MRI brain detected selective fronto-temporal atrophy, enlarged peri-Rolandic CSF spaces and hyper-intense signals in the caudate and lentiform nuclei on T2 weighted images. The urinary organic acid profile was consistent with the diagnosis of GA-I. Despite therapy there was no appreciable symptomatic and neurodevelopmental improvement a year later. Case 4 An 18-month-old male child had a history of acute onset head lag and loss of milestones at 6 months of age. This was associated with generalized tonic-clonic seizures and the episode was preceded by fever. The child had been treated with intravenous antibiotics on the clinical suspicion of pyogenic meningitis. The CSF studies, however, were normal. The child was born of a consanguineous marriage at term following an uneventful antenatal course. The postnatal period was normal and all the milestones had been achieved at an appropriate age. The unexplained neuro-regression at 6 months prompted evaluation by MRI, which revealed prominent sub-arachnoid CSF spaces over the frontal and anterior portions of the temporal lobes. On clinical and radiological suspicion of GA I, treatment with riboflavin, carnitine and baclofen had been promply instituted and seizure control achieved with phenobarbitone. The urine examination by GC-MS had been normal on two occasions 6 months apart. When examined at 18 months his weight was 8.9 kg. There was no macrocephaly (head circumference 45 cm). The child had been seizure-free for the last 4 months. Social smile had been regained at 14 months and incomplete head control was present. The boy was able to grasp objects. Neurological testing detected hypertonia, dystonia, orofacial dyskinesias, brisk deep tendon reflexes, weak rooting response and a blocked Moro response. A repeat MRI at 1 year of age had demonstrated bilateral putaminal signal abnormalities. The fronto-temporal atrophy was no longer evident. Reanalysis of urine sample by GC-MS detected a trace excretion of glutarate. The Diagnosis of GA-I was confirmed in this patient by mass tandem spectroscopy. By 24 months, the weight had increased to 10 kg. The child was rolling over partially and transferring objects, but he continued to have recurrent febrile episodes accompanied by temporary head lag and increased dystonic movements. The MRI at 3 years did not show any progression of the putaminal lesion and no other additional abnormalities were noted. In the interim the mother conceived. Prenatal diagnosis from amniotic fluid was success-fully attempted. The pregnancy was elect-ively terminated as the fetus was judged to be affected on the basis of raised levels of glutaric acid in the amniotic fluid. Discussion Glutaric acid and 3-hydroxyglutaric acid are the intermediary metabolities in the degradation pathway of lysine, hydroxylysine and tryptophan. In GA-I, the deficiency of the enzyme Glutary1 CoA dehydrogenase (GCDH) results in excessive accumulation of these metabolities in various tissues and body fluids. The disease is inherited as an autosomal recessive trait and mutations of the GCDH gene on chromosome 19 have been implicated in the causation of GA-I(1). GA-I has protean manifestations. Macrocephaly, a constant feature, is evident at birth and in the neonatal period(1-4) but may become less evident with progressive loss of cerebral volume(3-4). The micro-cephaly in our patients was presumably due to severe cerebral atrophy with advanced disease. It has not been reported previously in cases with GA-I. The usual age of presentation for GA-I is 6 months to 2 years of life(1-5). Acute neuroregression or dystonia following an initial phase of normal or almost normal development is a common mode of presentation, at times preceded by seizures(1,3,5). Antecedent events incrimi-nated as precipitating factors include febrile illness, immunization, minor head trauma and starvation(1,4). Metabolic derangement, which is the hallmark of organic acidurias, is minimal or absent even during acute symptomatic episodes(4). Hence, such cases are diagnosed as acute encephalitis, Reye syndrome or vaccine-induced encephalo-pathy(1,4). Presence of subdural or retinal bleeds in patients with GA-I even leads to a mistaken diagnosis of accidental injury or child abuse(1,6,7). In this manner the disorder is entirely overlooked and the stage is set for progressive necrosis of the caudate nucleus and putamen during subsequent acute metabolic crises resulting in a permanent neurologic handicap characterized by an extra-pyramidal movement disorder typical of GA-I(1,4,7). The other frequent pre-sentation of GA-I is a chronic encephalo-pathy associated with choreoathetosis or dystonia(8) often misdiagnosed as athetoid cerebral palsy(1). Report of asymptomatic adults with biochemical abnormalities characteristic of GA-I adds to the clinical variability with which the disease can present(4). Even when characteristic features of GA-I are present, the disorder can remain undignosed. This is not only because the disorder is rare and its awareness is far from optimum but also because the abnormal metabolites are detected in the urine intermittently or not at all even during acute metabolic crisis(1,7,9) as was observed in Case 4. These cases may represent a subtype having specific mutations correlating with the presence of significant residual enzyme activity(1). In such patients, GCDH assay from cultured fibroblasts provides the most accurate diagnostic modality (7,8). Stable isotope dilution technique is another useful diagnostic method that quantifies various metabolites including 3-hydroxygluta-rate(1,7). Neuroimaging serves as a useful tool, many a times providing the first clue to the diagnosis. Moreover, the findings on imaging correlate with the stage of the disease and can even be used to judge response to therapy(3,4,8,10). MRI is the imaging modality of choice. The earliest feature is fronto-temporal atrophy beginning in the second half of gestation and is also seen in the asymptomatic phase as well but progresses as symptoms develop(4,11). Basal ganglia abnormalities (volume loss or hyper-intensity on T2-weighted images), the incomplete opercularization of the insular cortex, widening of the sylvian fissures and CSF spaces anterior to the temporal lobes are characteristic of GA-I and are encountered in over 90% of patients with this disorder. The early changes in the white matter include delayed myelination and periventricular hypodensities. These are followed by diffuse attenuation and generalized atrophy in the late stages of the disease(1-4,11,12). The diagnosis of GA-I rests on the demonstration of urinary excretion of glutaric acid, 3-hydroxyglutaric acid and glutaconic acid by GC-MS(4,7). The detection of glutaric acid alone is insufficient for the diagnosis of this disorder. Metabolic disorders such as Glutaric acidemia type II, respiratory chain disorder, branched chain organic acidurias as well as riboflavin deficiency and valproate therapy have been associated with increased urinary glutarate excretion(7). For this reason detection of 3-hydroxyglutarate is considered the most specific(1,7,13). The understanding of pathogenesis of neuronal damage is crucial for the institution of various therapeutic modalities. Glutaric acid and 3-hydroxyglutaric acid are struc-turally similar to glutamate, a well-known excitotoxic amino acid. The intermediary metabolities stimulate the NMDA 2b receptors. The fetal brain is susceptible to injury as it has a preponderance of these receptors. Thus, the damage begins in utero, itself(1,4,14,15). During the encephlopathic crises there is massive activation of glutaminergic neurons from the cortex to putamen via caudate nucleus. Cellular energy depletion and low levels of inhibitory neurotransmitter GABA may also contribute to the neuropathogenesis(11,13). The man-agement should aim at early diagnosis, prevention of acute episodes and emergency care during intercurrent illnesses. Providing a low protein diet restricted in lysine and tryptophan, administration of pharmaco-logical doses of riboflavin and supplementa-tion of L-carnitine form the mainstay of therapy. Excessive restriction of tryptophan should, however, be avoided as it can precipitate irritability and sleep distur-bances(4). Synthetic formulae are currently not easily available in India. Administration of therapeutic doses of riboflavin is useful as the flavoprotein GCDH requires riboflavin as a cofactor. L-carnitine supplementation corrects the severe secondary carnitine deficiency that is associated with GA-I. In the presymptomatic patients, It can prevent the onset of symptomatic disease and is especially vital during the acute encephalo-pathic crises or in the older patient in whom dietary restriction is relaxed(1,2,4,11). Baclofen, a GABA analog is used to alleviate neurological symptoms(4,13). Anticonvul-sant therapy may be required if seizures are present. Early institution of therapy can provide gratifying results. It halts the further progression of the neurological disability in most patients. Its institution in the pre-symptomatic phase is known to prevent the onset of symptoms and may lead to normal development(1,4,11,13). As with other inherited disorders, genetic counselling and prenatal diagnosis are important aspects of management(11,13). There are no reports of cases of GA-I diagnosed and reported from India. The disorder may not be extremely rare considering the fact that four cases could be diagnosed in a short span of one year at our center. It is necessary for the pediatricians to be aware of this disorder so that neuroradiological and relevant biochemical tests are undertaken and cases are not misdiagnosed as encephalitis or cerebral palsy. Early detection is of utmost importance as prompt institution of therapy can limit and to some extent even reverse neurological handicap. Features such as precipitation of a significant neurological disease by febrile illnesses, worsening of neurological condition with subsequent episodes of illness and predominantly extra-pyramidal manifesta-tions should arouse a suspicion of the presence of a neurometabolic disease such as GA-I. Acknowledgement The authors are grateful to Professor M. Duran of University Children’s Hospital, Netherlands for analyzing urine samples of these patients.
Contributors:
All the authors were involved in patient management and drafting of the
manuscript. MNM will act as guarantor for the paper.
References
|
|
|