|
|
|
Indian Pediatr 2017;54: 955-960 |
 |
Growth Hormone Deficiency in Children: From
Suspecting to Diagnosing
|
|
Varuna Vyas, #Anil
Kumar and #Vandana Jain
From Department of Pediatrics,AIIMS, Jodhpur; and
#Division of Pediatric Endocrinology, Department of
Pediatrics, AIIMS, New Delhi; India.
Correspondence to: Dr Vandana Jain, Professor,
Division of Pediatric Endocrinology, Department of Pediatrics, All India
Institute of Medical Sciences, NewDelhi 110029.
Email: [email protected]
Received: May 22, 2017;
Initial Review: June 22, 2017;
Accepted: September 07, 2017.
|
|
Isolated Growth hormone deficiency is
an important and treatable cause of short stature. However, it is often
difficult to diagnose the condition with certainty due to the lack of a
single robust diagnostic test. Short children, other than those with the
classical phenotype of immature chubby facies, truncal obesity and
micropenis in boys, or those with history of cranial lesions with known
association with hypopituitarism, should be evaluated for growth hormone
deficiency only after excluding the other more common conditions. These
children typically have height markedly below that expected for their
midparental height with low height velocity and delayed bone age. Growth
hormone levels should be checked by provocative testing, after ensuring
that the child is euthyroid, and after priming with sex steroids if
indicated. Low levels of Insulin-like growth factor 1 and Insulin-like
growth factor binding protein 3 and pituitary abnormalities on
neuroimaging provide important corroborative evidence to the diagnosis.
Keywords:Diagnosis,Short stature,Height
velocity, Hypopituitarism, IGF1.
|
|
S
hort stature or poor growth is a common reason
for referral to a pediatrician. For children in whom causes such as
nutritional deficit, familial short stature, constitutional delay of
growth and puberty, chronic systemic illness or malabsorption (including
celiac disease), hypothyroidism, and in case of girls, Turner syndrome
have been excluded, the deficiency of growth hormone needs to be
considered.
Growth hormone (GH) is a polypeptide hormone secreted
by the anterior pituitary gland and is the chief driver of statural
growth during childhood. Human Growth Hormone (hGH) extracted from
cadaver pituitary glands was first isolated in 1956, and by 1959 its
clinical use in patients with presumed GH deficiency had started. In
1985, the use of hGH was abruptly halted amidst reports of death due to
Creutzfeldt Jacob Disease (CJD) in some recipients [1]. Later in the
same year, synthetic recombinant human growth hormone (rhGH) was
approved by the United States Food and Drug Administration (USFDA) for
clinical use. The indications for GH therapy were sequentially expanded
to include chronic renal insufficiency in 1993, Turner syndrome in 1997,
Prader Willi syndrome in 2000, small for gestational age in 2001,
idiopathic short stature in 2003, and Noonan syndrome in 2007 [2].
However, the unlimited availability of rhGH increased
the potential for its misuse; as an anabolic agent by athletes, as a
‘supposed’ anti-ageing agent, and in children who are not short by
definition but falling short on parents’ expectation of stature. Lack of
a simple one point robust test for diagnosing GH deficiency, compounds
this problem further [3]. Erroneous interpretation of a random GH level
as low by a physician leaves room for unwarranted prescriptions.
We, herein, present a brief review of the physiology
of growth hormone, and discuss the clinical, biochemical, radiological
and genetic investigations for diagnosis of growth hormone deficiency in
children, with special focus on avoidance of over-diagnosis of this
condition.
Physiology of Growth Hormone
GH is secreted from the anterior pituitary in a
pulsatile manner under hypothalamic regulation and other physiologic
regulators. There are approximately 10 daily pulses of GH secretion of
about 90 minutes duration in total, each separated by about 128 minutes.
The level of GH in between the pulses is nearly undetectable. Therefore
single isolated unprovoked GH measurement is of no value, and diagnosis
of GH deficiency is made by drawing 4-5 blood samples at half-hourly
intervals after administering a pharmacological stimulant [4].
Around 50% of the circulating GH is bound to Growth
hormone binding protein (GHBP). GH acts on the liver, muscle and bone,
and mediates the production and release of insulin-like growth factor
(IGF) 1 and 2 from these organs. Stimulation of linear growth in
children is chiefly mediated by IGF1. IGF2 is important for antenatal
growth but its role postnatally is not clear. Hepatic IGF1 circulates in
blood almost completely bound to IGF binding proteins (IGFBPs), a group
of sixstructurally related proteins that bind IGFs with high affinity.
Of these, IGFBP3 binds 75% to 90% of the circulating IGF1. This complex
is stabilized by Acid Labile Subunit (ALS), which increases the half
life of IGF1 [5].
Etiology of Growth Hormone Deficiency
The major congenital and acquired causes of GH
deficiency are summarized in Table I [6].
|
TABLE I Etiology of Growth Hormone Deficiency |
|
Congenital
Genetic
• Multiple pituitary hormone
deficiencies: Mutations in HESX1, LHX3, LHX4, SOX3, GLI2,
PROP1, PITX2 and PIT1genes.
• Isolated GH deficiency: Mutations in
GH1, GHRH and GHRHR genes
Congenital cranial malformations
• Holoprosencephaly, schizencephaly,
septo optic dysplasia
• Syndromic: Pallister Hall syndrome, Rieger
syndrome, Prader Willi syndrome
Acquired
Tumors
• Benign: Craniopharyngioma, arachnoid cyst,
pituitary adenoma, Rathke’s cleft cyst
• Malignant: Dysgerminoma, meningioma, glioma,
metastatic Hodgkin’s disease
Trauma: Surgical, skull fracture, birth
injury
Inflammation: Histiocytosis, sarcoidosis,
tuberculosis, meningitis, hemochromatosis, autoimmune
hypophysitis,
Pituitary apoplexy
Irradiation
|
Suspecting Growth Hormone Deficiency
Short stature is defined as height less than -2
standard deviations (SD) for age-and sex- appropriate population norms.
This implies that one in every 40 normal children is short. However, the
estimated prevalence of GH deficiency is 1 in 4,000 to 1 in 10,000 [7],
and hence, GH deficiency is a relatively rare cause of short stature.
Before evaluating a short child for GHD, commoner causes such as
physiological (familial short stature or constitutional delay of growth
and puberty), hypothyroidism, small for gestational age (SGA), chronic
systemic disease, celiac disease, Turner syndrome, or skeletal dysplasia
need to be considered and appropriately ruled out [7].
Children with GHD have normal anthropometry at birth.
Those with congenital hypopitutarism may have hypoglycemia, jaundice and
micropenis in the neonatal period. The most common presentation is with
complaint of poor growth in childhood. Typically, these children have
immature facies, mid-facial hypoplasia, pot belly, low height velocity
(<4 cm/ year during childhood) and delayed skeletal maturation.Presence
of midline anomalies such as single central incisor may be a pointer to
hypopituitarism. Acquired deficiency due to cranial lesions typically
presents as slowing or even cessation of linear growth. This may be
accompanied by other features such as polyuria and polydipsia (posterior
pituitary involvement), visual impairment and headache.
As per the consensus guidelines of the Growth Hormone
Research Society [7], investigation for GH deficiency should be
considered only in children who fulfil at least one of the criteria
listed in Box 1.
|
Box 1
Criteria for Considering Investigation for GH Deficiency |
|
• Height below -3 standard deviations (SD) for age-and
sex-appropriate population norms
• Height more than 1.5 SD below
midparental height
• Height below -2 SD and height velocity
below -1 SD for age and sex, or a decrease in height SD of
more than 0.5 over 1 yr in children aged >2 yr
• Children who are not short, but have
height velocity below -2 SD over 1 yr, or below -1.5 SD
sustained over 2 years (can occur in organic acquired GHD)
• Signs suggestive of intracranial lesion
• Features of multiple pituitary hormone
deficiencies
• Neonatal symptoms and signs of growth hormone deficiency
|
Diagnosis of Growth Hormone Deficiency
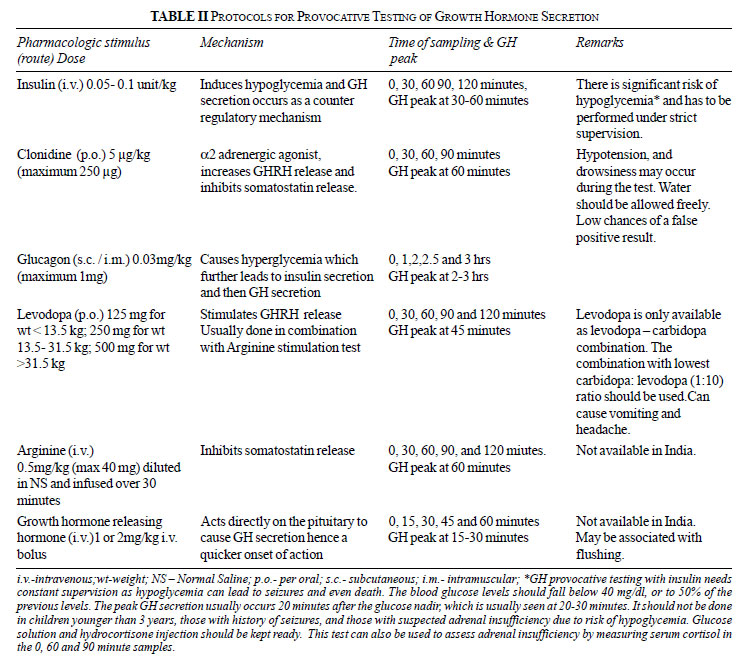
Provocative GH Testing
A period of fasting is required before all
provocative GH testing protocols. It is imperative to ensure that all
patients are euthyroid at the time of testing. A summary of the various
protocols is given in Table II [4]. It is recommended that
in children with suspected isolated GH deficiency, two provocative tests
should be done either sequentially or on separate days, and the
deficiency should be diagnosed only if the peak GH values in both the
tests are below the diagnostic cut-off.
Puberty and its implications on GH testing
With the onset of puberty, there is a large increase
in the concentration of circulating sex steroids, which augment GH
secretion. In the immediate prepubertal period, discriminating between
constitutional delay in growth and puberty (CDGP) and GHD is difficult.
In a study in 84 children, it was observed that 61% of the children with
Tanner stage I failed to achieve peak GH levels of >7 µg/L in response
to pharmacological provocative testing, while all the children at stage
IV/V were able to achieve peak GH >7µg/L. Administration of estrogen to
prepubertal subjects resulted in an increase in the range of peak GH
level from 1.9-20.3 µg/L to 7.2–40.5 µg/L [8]. In another study, 84 pre-
or early-pubertal boys with short stature, height velocity <4 cm/year
and failed GH provocative test were divided into two groups, one primed
with low dose (62.5 mg/m 2)
and the other, with conventional dose (125 mg/m2)
intramuscular testosterone. On retesting, 54% of the low dose group and
60% of the conventional dose group achieved peak GH values >10 µg/L [9].
Surveys of pediatric endocrinologists indicate that
there is no standard practice for priming peripubertal children with sex
steroids [10]. The Pediatric Endocrine Society in its recent guidelines
on diagnosis and treatment of GH deficiency has recommended that
prepubertal boys >11years and prepubertal girls >10 years of age should
undergo priming, especially if their predicted adult height is within -2
SD of the reference population mean. Priming with sex steroids reduces
the chances of misdiagnosis of children with constitutional delay of
growth and puberty as GHD [11]. Estradiol valerate1-2 mg can be used for
both boys and girls on each of the two evenings prior to the test.
Alternatively, boys can be primed with intramuscular depot testosterone
50 to 100 mg 1 week prior to the test [11].
Peak GH during provocative testing: what is the
cut-off for GH deficiency?
Conventionally peak GH values below 7 µg/L or 10 µg/L
after provocative tests are considered as indicative of GH deficiency.
However, these cut-offs are a matter of debate. In a study that
evaluated the secretion of GH after stimulation by clonidine, insulin
and arginine in 7- to -18 year-old children with normal stature, it was
noted that the mean (SD) peak values for GH were higher in response to
clonidine [21.0 (10.7) µg/L], compared to those in response to arginine[13.1
(6.1) µg/L] or insulin [14.2 (6.3) µg/L] [12]. It was also observed that
the mean (SD) peak GH level after clonidine stimulation increased from
12.8 (5.1) µg/L in Tanner I girls to 35.5 (5.1) µg/L in Tanner IV/V
girls and similarly, from 16.9 (6.7)µg/L in Tanner I boys to 26.5 (12.0)
µg/L in Tanner IV/ V boys. In 1996, Ghigo,et al. [13] published a
study that assessed the reliability of some of the provocative agents
used in GH testing. Ideally, administration of these agents should be
able to produce peak GH values above the conventional cut-offs in normal
children. In 472 children, including those with short as well as normal
stature, but with normal height velocity, normal IGF1 levels, and no
delay in bone age, it was observed that using various provocative
agents, 23% to 49% of these GH non-deficient subjects failed to achieve
peak GH values above 10 µg/L, and 10–24% had peak GH <7 µg/L [13].
The Pediatric Endocrine society (PES) 2016 guidelines
also caution against using provocative GH testing as the sole basis for
diagnosing GH deficiency [11]. While patients with severe GH deficiency
generally have very low values on provocative testing, the threshold
that distinguishes partial GHD from normal is not clear. It is also
important to remember that obese children have blunted GH peak in
response to various stimuli [11]. Data from several big post-marketing
surveys of GH therapy suggest that the most significant increase in
height velocity and height standard deviations are seen in those
children who had a peak GH value <5µg/L at diagnosis [14-16].
Situations when provocative GH testing is not needed
for diagnosing GH deficiency
In short children with low height velocity, who have
a known hypothalamic-pituitary defect, such as a major congenital
malformation, history of cranial tumor or irradiation, and deficiency of
at least one more pituitary hormone, provocative GH testing is not
needed to diagnose GHD. Similarly, a newborn with hypoglycemia, who has
a GH level <5 µg/L in a critical blood sample and has the classical
triad of hypoplastic anterior pituitary, ectopic posterior pituitary and
abnormal stalk on neuroimaging; and/or deficiency of at least one other
pituitary hormone can be diagnosed as having GH deficiency [11].
IGF1 and IGFBP3
The production of IGF1 and IGFBP3 is dependent on GH.
More than 75% of circulating IGF1 is bound to IGFBP3 and this complex
has a half life of 16 hours [17]. The two, hence, seem to be very
convenient molecules that can be measured for a functional bioassay of
GH. However, these also have their own limitations; the foremost being
that their levels may be affected by nutritional status, age, thyroid
function, degree of sexual maturation, genetic factors and liver
function [18]. IGFBP3 is less affected by these factors as compared to
IGF1. There is no single cut-off for IGF1 and IGFBP3, and values have to
be compared to age, genderand pubertyspecific normative reference data
[19-21].
In a retrospective analysis in 33 children with GH
deficiency (diagnosed on the basis of short stature, delayed bone age,
two failed GH provocative tests, hypothalamo-pituitary anomalies on MRI,
height catch-up on GH therapy, and a repeat GH provocative test value
<10 µg/L after completion of linear growth) and 56 children with
idiopathic short stature, a cut-off value of <10 µg/L to define GH
deficiency using provocative tests was 100% sensitive and 57% specific.
Decreasing the cut-off to 7 µg/L changed the sensitivity and specificity
to 66% and 78%, respectively. Low IGF1 had a sensitivity of 73% and
specificity of 95%, low IGFBP3 had a sensitivity of 30% and specificity
of 98%, andlow height velocity had a sensitivity of 82% and specificity
of 43%. Combination of height velocity and IGF1 had a sensitivity of 95%
and specificity of 96% [22].In another similar study, IGFBP3 and IGF1
were evaluated in patients with GH deficiency and idiopathic short
stature. It was observed that for diagnosing GHD, low IGF1 (<5th centile)
had a sensitivity of 69%. The specificity was 91% in children aged <11
years but only 53% in children aged >11 years.The specificity of low
IGFBP3 was 100% for diagnosis of GHD, but sensitivity was less than 50%
[23].
In conclusion, low levels of IGF1 and IGFBP3 are
reasonably specific for diagnosis of GHD, especially in children younger
than 10-11 years of age, but their sensitivity is low, so that normal
values are not sufficient to exclude GH deficiency. If height velocity
is combined with the laboratory parameters, the sensitivity and
specificity of the diagnosis improve.
Neuroimaging
As per Growth Hormone Research Society Consensus
Guidelines, in patients with confirmed isolated GH deficiency or
Multiple Pituitary Hormone Deficiency, magnetic resonance imaging (MRI)
with 2 mm slices should be done to note the height/volume of anterior
pituitary, position of posterior pituitary and anatomy of the stalk. The
abnormalities include pituitary hypoplasia (gland height <-2SD for age),
ectopic posterior pituitary and stalk abnormalities [7].
In a recent study in 68 children diagnosed with GH
deficiency before 4 years of age, MRI abnormalities were noted in 84%
patients with isolated GH deficiency, of which 49% had only isolated
pituitary hypoplasia; while in patients with multiple hormone
deficiencies100% had complex defects [24].
Genetic Studies
Genetic mutations are identified in a relatively
small number of patients with GH deficiency. However, a genetic
diagnosis should be established where feasible in children with
congenital GH deficiency. It helps in planning follow-up and appropriate
evaluation of other family members.GH deficiency can occur due to
mutations in transcription factors genes involved in the development of
the pituitary gland (summarized in Web Table I), which
typically are associated with multiple pituitary hormone deficiencies;
or due to mutations in genes encoding growth hormone (GH1),
growth hormone releasing hormone (GHRH) or its receptor (GHRHR),
which lead to isolated GH deficiency [25-27].
In a study by Desai, et al. [28] in 97
patients with isolated GHD, GH1 gene deletion was noted in 17%,
while GHRHR gene mutations were present in 35%. In recent studies
from Delhi, among 51 patients with multiple pituitary hormone
deficiency, 6% had mutations in PROP1 gene and 14% in POU1F1
gene [29]; while among 116 patients with isolated GH deficiency,
mutations in GH1 and GHRHR genes were observed in 7% and
21% patients, respectively [30].
Conclusion
The key messages related to diagnosis of GH
deficiency in children are summarized in Box 2.
BOX 2
Key Messages Related to Diagnosis of Growth Hormone
Deficiency
|
|
• Growth hormone deficiency is a relatively
rare cause of short stature, and commoner causes should be ruled
out before testing.
• Children with growth hormone deficiency
typically have height more than 2 SD below the population norm, low
height velocity, delayed bone age, immature facies, midfacial
hypoplasia and trunkal obesity.
• Provocative testing lacks specificity.
Depending on the provocative agent, between 10-50% of normally
growing children can have peak growth hormone values below 10 µg/L.
Hence, these tests should be undertaken only in those children where
the pre-test probability is already high, such as those fulfilling
at least one of the criteria listed in Box 1.
• Prior to provocative testing, ensure a period
of fasting, euthyroid status and appropriate sex steroid priming.
• Low levels of IGF 1 and IGFBP3, especially in
combination with low height velocity are important pointers to
growth hormone deficiency.
• Presence of structural anomalies of the
pituitary strengthens the diagnosis, and MRI is recommended in all
patients diagnosed with growth hormone deficiency.
• Genetic testing should be done in children with congenital
growth hormone deficiency, where feasible.
|
Contributors: The review was conceptualized by
VJ. All thethree authors contributed to literature search and writing
the review.
Funding: None; Competing interests: None
References
1. Tattersall R. A history of growth hormone. Horm
Res. 1996;46:236-47.
2. Ayyar VS. History of growth hormone
therapy. Indian J Endocrinol Metab. 2011;15:S162-S165.
3. Guyda HJ. Growth hormone testing and the short
child. Pediatr Res. 2000;48:579-80.
4. Carrillo AA,Bao Y. Hormonal dynamic tests and
genetic tests used in pediatric endocrinology. In: LifshitzF,
editor. Pediatric Endocrinology, 5thed. New York: Informa Healthcare;
2007.p.737-67.
5. Domené HM, Bengolea SV, Martínez AS, Ropelato MG,
Pennisi P, Scaglia P, et al. Deficiency of the circulating
insulin-like growth factor system associated with inactivation of the
acid-labile subunit gene. N Engl J Med. 2004;350:570-7.
6. Rosenbloom AL, Connor EL. Hypopituitarism and
other disorders of the growth hormone–insulin-like growth factor-I axis.
In: LifshitzF, editor. Pediatric Endocrinology. 5thed. New York:
Informa Healthcare; 2007.p.65-99.
7. Growth Hormone Research Society. Consensus
guidelines for the diagnosis and treatment of growth hormone (GH)
deficiency in childhood and Adolescence: Summary statement of the GH
research society. J Clin Endocrinol Metab. 2000;85:3990-3.
8. Marin GA, Domene HM, Barnes KM, Blackwell BJ,
Cassorla FG, Cutler GB Jr. The effects of estrogen priming and puberty
on the growth hormone response to standardized treadmill exercise and
arginine-insulin in normal girls and boys. J Clin Endocrinol Metab.
1994;79: 537-41.
9. Gonc EN, Yordam N, Kandemir N, Alikasifoglu A.
Comparison of stimulated growth hormone levels in primed versus unprimed
provocative tests. The effect of various testosterone doses on growth
hormone levels. Horm Res. 2001;56:32–7
10. Murray PG, Dattani MT, Clayton PE. Controversies
in the diagnosis and management of growth hormone deficiency in
childhood and adolescence. Arch Dis Child. 2016;101:96-100.
11. Grimberg A, DiVall SA, Polychronakos C, Allen DB,
Cohen LE, Quintos JB, et al. Guidelines for growth hormone and
insulin-like growth factor-I treatment in children and adolescents:
Growth hormone deficiency, idiopathic short stature, and primary
insulin-like growth factor-I deficiency. Horm Res Paediatr.
2016;86:361-97.
12. Zadik Z, Chalew SA, Kowarski A. Assessment of
growth hormone secretion in normal stature children using 24-hour
integrated concentration of GH and pharmacological stimulation. J Clin
Endocrinol Metab. 1990;71:932–6.
13. Ghigo E, Bellone J, Aimaretti G, Bellone S, Loche
S, CappaM,et al. Reliability of provocative tests to assess
growth hormone secretory status. Study in 472 normally growing children.
J ClinEndocrinolMetab. 1996;81: 3323-7.
14. Ranke MB, Lindberg A, Chatelain P, Wilton P,
Cutfield W, Albertsson-WiklandK, et al. Derivation and validation
of a mathematical model for predicting the response to exogenous
recombinant human growth hormone (GH) in prepubertal children with
idiopathic GH deficiency. J Clin Endocrinol Metab. 1999;84:1174-83.
15. Bright GM, Julius JR, Lima J, Blethen SL. Growth
hormone stimulation test results as predictors of recombinant human
growth hormone treatment outcomes: preliminary analysis of the national
cooperative growth study database. Pediatrics. 1999;104:1028-31.
16. Fujieda K, Hanew K, Hirano T, Igarashi Y, Nishi
Y, TachibanaK, et al. Growth response to growth hormone therapy
in patients with different degrees of growth hormone deficiency. Endocr
J. 1996;43:S19-S25.
17. Clemmons DR. Role of insulin-like growth factor-I
in diagnosis and management of acromegaly. Endocr Pract. 2004;10:362-71.
18. Cianfarani S, Liguori A, Boemi S, Maghnie M,
Iughetti L, WasniewskaM,et al. Inaccuracy of insulin-like growth
factor (IGF) binding protein (IGFBP)-3 assessment in the diagnosis of
growth hormone (GH) deficiency from childhood to young adulthood:
association to low GH dependency of IGF-II and presence of circulating
IGFBP-3 18-kilodalton fragment. J Clin Endocrinol Metab. 2005;90:
6028-34.
19. Dehiya RK, Deepa B, Chhaya K, Desai MP. Insulin
like growth factor-I, insulin like growth factor binding protein-3 and
acid labile subunit levels in healthy children and adolescents residing
in Mumbai suburbs. Indian Pediatr. 2000;37:990-7.
20. Lofqvist C, Andersson E, Gelander L, Rosberg S,
Blum WF, Wikland KA. Reference values for IGF-I throughout childhood and
adolescence: Amodel that accounts simultaneously for the effect of
gender, age, and puberty. J Clin Endocrinol Metab. 2001;86:5870-6.
21. Lofqvist C, Andersson E, Gelander L, Rosberg S,
Hulthen L, Blum WF, et al. Reference values for insulin-like
growth factor-binding protein-3 (IGFBP-3) and the ratio of insulin-like
growth factor-I to IGFBP-3 throughout childhood and adolescence. J Clin
Endocrinol Metab. 2005;90:1420-7.
22. Cianfarani S, Tondinelli T, Spadoni GL, Scirè G,
Boemi S, Boscherini B. Height velocity and IGF I assessment in the
diagnosis of childhood onset GH insufficiency: Do we still need a second
GH stimulation test? J Clin Endocrinol Metab. 2002;57:161-7.
23. Cianfarani S, Liguori A, Boemi S, Maghnie M,
Iughetti L, WasniewskaM,et al. Inaccuracy of insulin-like growth
factor (IGF) binding protein (IGFBP)-3 assessment in the diagnosis of
growth hormone (GH) deficiency from childhood to young adulthood:
association to low GH dependency of IGF-II and presence of circulating
IGFBP-3 18-kilodalton fragment. J Clin Endocrinol Metab. 2005;90:
6028-34.
24. Pampanini V, Pedicelli S, Gubinelli J, ScirèG,
Cappa M, BoscheriniB,et al. Brain magnetic resonance imaging as
first-line investigation for growth hormone deficiency diagnosis in
early childhood. Horm Res Paediatr. 2015;84:323-30.
25. Alatzoglou KS, Dattani MT. Genetic causes and
treatment of isolated growth hormone deficiency—an update. Nat Rev
Endocrinol. 2010;6:562-76.
26. Mullis PE. Genetic control of growth. Eur J
Endocrinol. 2005;152:11-31.
27. Online Mendelian Inheritance in Man, OMIM (TM).
McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University
(Baltimore, MD) and National Center for Biotechnology Information,
National Library of Medicine (Bethesda, MD). Availablefrom:http://www.ncbi.nlm.nih.gov/omim/.
Accessed January 01, 2017.
28. Desai MP, Mithbawkar SM, Upadhye PS, Rao SC,
Bhatia V, Vijaykumar M. Molecular genetic studies in isolated growth
hormone deficiency (IGHD). Indian J Pediatr. 2013;80:623-30.
29. Birla S, Khadgawat R, Jyotsna VP, Jain V, Garg
MK, Bhalla A, et al. Identification of novel PROP1 and
POU1F1mutations in patients with combined pituitary hormone
deficiency. Horm Metab Res. 2016;48:822-7.
30. Birla S, Khadgawat R, Jyotsna VP, Jain V, Garg
MK, Bhalla AS, et al. Identification of novel GHRHR and
GH1 mutations in patients with isolated growth hormone deficiency.
Growth Horm IGF Res. 2016;29:50-6.
|
|
|
 |
|