|
|
|
Indian Pediatr 2016;53: 977-982 |
 |
Phenotype
of Dent Disease in a Cohort of Indian Children
|
|
Swati Bhardwaj, Ranjeet Thergaonkar, Aditi Sinha,
Pankaj Hari, * HI Cheong and
Arvind Bagga
From the Department of Pediatrics, AIIMS, New Delhi,
India; and *Department of Pediatrics, Research Coordination Center for
Rare Diseases, and Kidney Research Institute, Seoul National University
College of Medicine, Seoul, Korea.
Correspondence to: Prof Arvind Bagga, Division of
Nephrology, Department of Pediatrics, All India Institute of Medical
Sciences, Ansari Nagar, New Delhi 110 029, India.
Email: [email protected]
Received: March 07, 2016;
Initial review: May 19, 2016;
Accepted: September 01, 2016.
.
|
Objective: To describe the clinical and genotypic features of Dent
disease in children diagnosed at our center over a period of 10 years.
Design: Case series.
Setting: Pediatric Nephrology Clinic at a
referral center in Northern India.
Methods: The medical records of patients with
Dent disease diagnosed and followed up at this hospital from June 2005
to April 2015 were reviewed. The diagnosis of Dent disease was based on
presence of all three of the following: (i) low molecular weight
proteinuria, (ii) hypercalciuria and (iii) one of the
following: nephrolithiasis, hematuria, hypophosphatemia or renal
insufficiency, with or without mutation in CLCN5 or OCRL1
genes.
Results: The phenotype in 18 patients diagnosed
with Dent disease during this period was characterized by early age at
onset (median 1.8 y), and polyuria, polydipsia, salt craving,
hypophosphatemic rickets and night blindness. Rickets was associated
with severe deformities, fractures or loss of ambulation in six
patients. Nephrocalcinosis was present in three patients, while none had
nephrolithiasis. Generalized aminoaciduria was seen in 13 patients, two
had glucosuria alone, and one had features of Fanconi syndrome. Over a
median follow up of 2.7 years, one patient developed renal failure.
Genetic testing (n=15) revealed 5 missense mutations and 3
nonsense mutations in CLCN5 in 13 patients. Five of these
variations (p.Met504Lys, p.Trp58Cys, p.Leu729X, p.Glu527Gln and
p.Gly57Arg) have not been reported outside the Indian subcontinent.
Conclusion: Our findings suggest a severe
phenotype in a cohort of Indian patients with Dent disease.
Keywords: CLCN5, Hypophosphatemic rickets, Night blindness,
Polyuria.
|
|
Dent disease is an X-linked disorder of proximal
tubular function characterized by low molecular weight proteinuria, the
most consistent feature, as well as hypercalciuria, nephrocalcinosis,
nephrolithiasis and progressive renal failure [1,2]. The condition
presents in boys during early childhood with symptoms of renal stones
(pain abdomen, hematuria), bone pains or deformities due to rickets or
with incidentally detected low molecular weight proteinuria. Progression
to end stage renal disease (ESRD) occurs between 3rd to 5th decades in
30-80% of affected males.
The disease is caused in 60% patients by inactivating
mutations in the CLCN5 gene, located on Xp11.22 encoding a 746
amino acid Cl -/H+
exchanger (Dent disease 1) [4]; 15% of patients show mutations in the
OCRL1 gene (Dent disease 2) on chromosome Xq25, which encodes
phosphatidylinositol 4,5-biphosphate 5-phosphatase [5,6]. There is
genetic heterogeneity and no genotype phenotype correlations are
established [6-8]. A previous report on three patients with Dent disease
from this center emphasized the early onset of symptoms and occurrence
of night blindness, presumably secondary to urinary wasting of retinol
binding protein (RBP) [9]. In this study, we now report our experience
on diagnosis and management of a cohort of 18 patients with the
condition, including follow-up of those reported previously.
Methods
The medical records of patients with Dent disease
diagnosed and followed up at this hospital from June 2005 to April 2015
were reviewed. Three patients diagnosed before this period were also
included. The diagnosis of Dent disease was based on presence of all
three of the following: (i) low molecular weight proteinuria,
defined as increased excretion of b2
microglobulin >1500 µg/L (normal <300 µg/L),
(ii) hypercalciuria (urinary calcium excretion >4 mg/kg/day or
calcium creatinine ratio, UCa/UCr
>0.2 mg/mg), and (iii) one of the following: nephrolithiasis,
hematuria, hypophosphatemia or renal insufficiency, with or without
mutation in CLCN5 or OCRL1 genes. Patients with other
causes of proximal tubular dysfunction (proximal renal tubular
acidosis), hypercalciuria (distal renal tubular acidosis, idiopathic
hypercalciuria), refractory rickets (familial hypophosphatemic rickets,
vitamin D dependence) and nephrolithiasis were excluded.
Clinical and biochemical details were recorded at
diagnosis and at follow up. Standard deviation score (SDS) for weight
and height were calculated using World Health Organization charts and
AnthroPlus v.1.0.4 calculator (www.who.int/growthref/tools/en). Blood
levels of calcium, phosphate, alkaline phosphatase, electrolytes,
creatinine, pH, bicarbonate, 25-hydro-xyvitamin D and parathormone were
measured and compared to age-appropriate cut-offs. Timed urinary
excretion of phosphate and creatinine was used to estimate tubular
maximum for phosphate reabsorption/glomerular filtration rate (TmP/GFR);
estimated glomercular filteration rate (eGFR) by the modified Schwartz
formula was used to classify stages of chronic kidney disease (CKD)
[10]. Height SDS, blood creatinine, eGFR and calcium excretion were
compared at presentation and follow-up by Wilcoxon signed rank test
(SPSS version 15.0, SPSS Inc., Chicago).
Genomic DNA, isolated from peripheral blood
leukocytes, was amplified by polymerase chain reaction (PCR) using
primers for all exons of the CLCN5 gene [11], followed by Sanger
sequencing. In case the CLCN5 sequence was negative for
variations, the OCRL1 gene was screened. Pathogenecity prediction
software, sorting intolerant from tolerant (SIFT,
http://sift.jcvi.org/) and PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/)
were used to predict the effect of novel exonic variations.
Results
Clinical and biochemical data on 18 patients with
Dent disease are summarized in Table I. Detailed data are
shown in Web Table I. All patients except two pairs
(maternal uncle and nephew Pt. 6, 7; brothers Pt.11, 12) were unrelated.
One patient had a history of similar illness in maternal uncle who had
end stage renal failure and renal transplantation at 38-years of age.
The median age at onset of symptoms was 1.8 years and that at diagnosis
was 8 years. All patients had short stature with median height SDS at
presentation of -3.6. The presenting features included bony deformities
due to rickets and complaints of polyuria, polydipsia and craving for
salty foods. One or more episodes of night blindness, manifested on
multiple occasions at variable periods after onset of symptoms in 12
patients (66.7%), were responsive to therapeutic doses of vitamin A. One
patient presented with persistent nephrotic range proteinuria without
edema; Dent disease was suspected based on the presence of rickets, low
molecular weight proteinuria and hypercalciuria. At presentation, the
median eGFR was 72 mL/min/1.73 m 2;
14 (77.8%) and 5 (27.8%) patients had eGFR <90 mL/min/1.73 m2
and <60 mL/min/1.73 m2,
respectively. Medullary nephro-calcinosis was
present in two patients at diagnosis and in one on follow up. None of
the patients had nephrolithiasis.
TABLE I Summary of Clinical and Biochemical Features of Patients with Dent Disease
|
Parameter |
Value |
|
Age at onset (y) |
1.8 (0.3,8) |
|
Age at diagnosis (y) |
8.0 (1.5,14) |
|
Height SDS at presentation |
–3.6 (-8.4,-1.9) |
|
Time from onset to diagnosis (y) |
4.0 (1,13) |
|
Polyuria, polydipsia, n (%) |
16 (88.9) |
|
Salt preference, n (%) |
9 (50.0) |
|
Rickets, n (%) |
18 (100) |
|
Night blindness, n (%) |
12 (66.7) |
|
Serum creatinine (mg/dL) |
0.6 (0.3,1.3) |
|
eGFR at presentation (mL) |
72 (28,126) mL/min/1.73 m2 |
|
Hypokalemia, n (%) |
13 (72.2) |
|
Serum phosphate (mg/dL) |
2.6 (2.4,3) dL |
|
TmP/GFR (mg/dL) |
1.7 (1.1,3.6) |
|
24-hr urine protein (mg) |
1150 (520,3400) |
|
24-hr urine calcium (mg/kg) |
8.1 (3, 20) |
|
Aminoaciduria, n (%) |
13 (72.2) |
|
Follow up duration (y) |
2.7 (0.3,20.6) y |
|
Height SDS |
–4.3 (-8.4, -1.5) |
|
eGFR (mL/min/1.73 m2) |
66 (28, 120) |
|
24-hr urine calcium (mg/kg) |
7 (2, 17.6) |
|
Values as median (range) unless specified otherwise. |
All patients had radiological evidence of rickets
with normal blood levels of calcium and alkaline phosphatase.
Hypophosphatemia was seen in 17 (94.4%) patients with median blood level
of phosphate 2.6 mg/dL; and TmP/GFR below 4 mg/dL in all. Serum
25-hydroxyvitamin D level was low (<30 ng/mL) in 5 of 12 patients
tested. Ten patients showed normal levels of parathormone and two each
with renal dysfunction and vitamin D deficiency had elevated levels.
Many of the patients had received therapeutic dose of vitamin D before
presenting to our center. We administered vitamin D only if the patient
showed low levels of 25-OD vitamin D. Rickets was refractory to
therapeutic doses of vitamin D in all patients. Additional abnormalities
of proximal tubular function were generalized aminoaciduria in 13
(72.2%), glucosuria in 2 and normal anion gap metabolic acidosis in 2
patients including one with features of Fanconi syndrome. All patients
received therapy with citrate and phosphate supplements; 3 also received
hydro-chlorothiazide for treatment of hypercalciuria for a brief
duration. Hypokalemia prompted discontinuation of the same.
Follow-up data were available for all, except one (Web
Table I). At a median (range) follow-up of 2.7 (0.3-20.6) years,
the median (range) height SDS was -4.3 (-8.4 to -1.5), similar to that
at diagnosis (P=0.81). Radiological healing of rickets occurred
in 9 of 12 patients with more than 6 months follow-up. Bony deformities
persisted and three patients underwent corrective osteotomy. The median
(range) eGFR at follow up, 66 (28-120) mL/min/1.73 m2,
was also similar to that at diagnosis (P=0.80). Seven patients,
including one with nephrocalcinosis showed decline in renal function; 4
showed >25% decline in eGFR. An additional patient had CKD stage IV at
9.5 years of age. Kidney biopsy in his sibling showed global sclerosis
in most glomeruli and significant interstitial fibrosis and tubular
atrophy. Hypercalciuria persisted at follow-up (median 7.0; range 2-17.6
mg/kg/day), without change from baseline (P=0.78).
Genetic analysis: Sequence analysis of the
CLCN5 gene in 15 patients revealed eight mutations in exons 3, 7, 9,
10, 11 and 12 in 13 patients (Web Table II, Fig. 1)
[12-15]. Five were missense mutations (p.Ser244Leu, p.Met504Lys,
p.Trp58Cys, p.Glu527Gln and p.Gly57Arg), and three were nonsense
mutations (p.Leu729X, p.Arg648X and p.Arg637X). Two patients did not
show mutations in the CLCN5 or OCRL1 genes. Both the novel
missense mutations (p.Glu527Gln and p.Gly57Arg) were predicted to affect
protein function by either SIFT or PolyPhen2 computer programs. All
three nonsense mutations including one novel mutation (p.Leu729X) were
expected to be pathogenic.
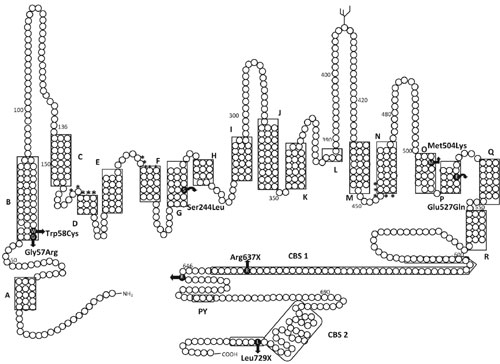 |
|
Fig. I Schematic representation of
ClC-5 mutations in 13 patients with Dent disease. The predicted
topology of the protein is drawn from Wu, et al. [14] and
Dutzler, et al. [15]. ClC-5 consists of 18 á helices, A to R,
which are indicated by the boxed areas. Mutations in ClC-5 are
shown as solid black circles with the resulting amino acid
change indicated besides.
|
Discussion
The present report describes a severe phenotype of
Dent disease. Most patients showed pathogenic variants involving the
CLCN5 gene. The median age at diagnosis was similar to a cohort of
117 European patients and pooled data from 377 patients [16]. However,
patients in this report showed relatively early onset of symptoms
compared to other series (Table II) [5,16-18]. While
rickets is reported in infants with Dent disease, early onset with
polyuria, polydipsia and night blindness is not described [1,12-19]. We
also report the occurrence of renal failure in the first decade of life
in patients with a common mutation in CLCN5, p.Ser244Leu.
TABLE II Phenotypic Findings in Patients with Dent Disease 1 (CLCN5 mutations) in the Present and Previous Reports
|
Features |
Hoopes [5]*, |
Sekine [17], |
Mansour-Hendili [16], |
Anglani [18], |
Present study, |
|
n=19 |
n=61 |
n=117 |
n=47 |
n=13 |
|
Median age at diagnosis (y) |
10 |
- |
7 |
- |
8 |
|
Low molecular weight proteinuria, % |
100# |
100# |
99.0 (n=111) |
97.9 |
100# |
|
Hypercalciuria, % |
100# |
46.0 (n=54) |
88.0 (n=99) |
91.5# |
100# |
|
Nephrocalcinosis, % |
89.5 (n=19) |
38.0 (n=53) |
61.0 (n=102) |
83.0 |
23.1 |
|
Nephrolithiasis, % |
29.4 (n=17) |
- |
33.0 (n=91) |
29.8 |
0 |
|
Renal insufficiency^, % |
26.3 (n=19) |
8.0 (n=53) |
47.0 (n=110) |
10.6 |
38.4 |
|
Rickets, % |
38.4 (n=13) |
0 (n=61) |
15.0 (n=93) |
36.2 |
100 |
|
Hypophosphatemia, % |
50.0 (n=18) |
- |
54.0 (n=76) |
36.2 |
92.3 |
|
Aminoaciduria, % |
75.0 (n=8) |
- |
61.0 (n=39) |
- |
76.9 |
|
Hypokalemia, % |
35.3 (n=17) |
- |
39.0 (n=86) |
- |
76.9 |
|
Metabolic acidosis, % |
- |
- |
13.0 (n=68) |
- |
7.7 |
|
Glucosuria, % |
38.9 (n=18) |
- |
40.0 (n=70) |
- |
15.4 |
|
Concentrating defect, % |
- |
- |
72.1 (n=43) |
- |
84.6 |
|
*Data pooled for patients with or without mutations in CLCN5
gene; #Considered an essential criterion for diagnosis;
ˆDefinition varies across reports; renal insufficiency was eGFR<60
mL/min/1.73 m2 (CKD stage III) in the present study; 1 patient
had eGFR <30 mL/min/1.73 m2 (CKD stage IV). |
Most series report rickets in up to one-third
patients with Dent disease 1 [5,16-18]. Wrong, et al. [2]
reported rickets in 40% patients and hypophosphatemia in one-third
patients, with satisfactory response to vitamin D therapy. All our
patients showed refractory rickets with hypophosphatemia in 94.4 %
cases. Similar to four of our patients, all nine affected boys from two
European families with the CLCN5 mutation p.Ser244Leu, in whom
the mutation was first described, showed rickets [20]. However, none of
the patients with this mutation from a large pedigree in southern United
States had features of rickets [21]. It is unclear whether these
phenotypic variations reflect differences in severity of coexistent
vitamin D deficiency, dietary and environmental factors, delayed
diagnosis, or effect of modifier genes.
Polyuria, an important symptom in the our patients,
is reported in Dent disease [2]. While formal water deprivation testing
was not performed, a defect in urinary concentration is likely, similar
to other inherited renal tubular disorders with secondary nephrogenic
diabetes insipidus [22]. This may be mediated by downregulation of
expression of aquaporin 2 via the calcium sensing receptor in
apical membrane of medullary collecting duct [22,23]. The high incidence
of polyuria in these patients might explain the low prevalence of
nephrolithiasis, the former serving as a physiological mechanism
preventing stone formation. Moreover, it is also reported that the
degree of hypercalciuria may not relate to development of
nephrocalcinosis or renal failure [24]. Despite high rates of
hypercalciuria and nephrocalcinosis (97.6% and 87.8%, respectively),
only 12% of 41 patients of Dent disease showed renal failure [18].
Additional genetic or environmental factors may contribute to the
occurrence of nephrocalcinosis/nephrolithiasis and consequent renal
dysfunction.
Vitamin A-responsive night blindness, first reported
in Dent disease in patients from our center [9], was observed in
two-thirds of the present patients, compared to 37.5% in another report
[25]. The condition is attributed to high urinary losses of RBP [3,25]
and reduced blood levels of retinol and RBP [25], but information on
dietary intakes and blood retinol levels is lacking. While insufficient
vitamin A intake might predispose patients with Dent disease to
clinically overt vitamin A deficiency, none of the unaffected family
members developed night blindness.
Previous studies have not attempted grading of CKD,
precluding comparisons in different cohorts. Of the 18 patients, 14
(77.8%) showed eGFR<90 mL/min/1.73 m 2
at presentation and 7 showed decline in eGFR on follow
up. One patient (Pt. 11) had CKD stage IV at the age of 9.5 years; his
younger sibling (Pt. 12) showed glomerular and tubulointerstitial
scarring on renal biopsy, emphasizing the risk of progression into renal
failure even in patients without nephrocalcinosis. This family with 3
affected boys with renal failure within first decade of life represents
a severe phenotype. While genetic testing showed a commonly described
mutation, p.Ser244Leu, the occurrence of renal failure in first decade
with this mutation is not reported.
We identified 5 missense and 3 nonsense mutations in
13 patients. Five of these 8 mutations (p.Met504Lys, p.Trp58Cys,
p.Leu729X, p.Glu527Gln and p.Gly57Arg) have not been reported outside
the Indian subcontinent, the small numbers preclude any
genotype-phenotype correlation. Of the 192 mutations of CLCN5
that are reported, approximately 17%, 36.5% and 28% are nonsense,
missense and frameshift mutations, respectively [16]. The missense
mutations p.Glu527Gln and p.Ser244Leu alter ClC-5
a-helices P and G
respectively, which interfere with dimer interface formation [14,15].
The mutation p.Met504Lys is expected to disrupt the function of helix O,
and p.Trp58Cys and p.Gly57Arg that of helix B, thereby reducing chloride
channel function; functional characterization has not been carried out
for these mutations.
The study is limited by small sample size,
retrospective design, and lack of genetic testing in asymptomatic family
members. However, there are important phenotypic differences from
previously reported cohorts, including low prevalence of
nephro-calcinosis and occurrence of CKD within first decade of life.
Apart from the usual features, Dent disease in this cohort of Indian
boys has a relatively severe phenotype with early onset of symptoms,
hypophosphatemic rickets and night blindness.
Contributions: SB, RT, AS, PH, AB:
diagnosis and management of patients; SB, RT: data collection; AS, PH,
AB: analytical inputs and review of literature; HC: performed the
sequencing of the genes. All authors participated in preparation
of the manuscript and approved the final version submitted for
publication.
Funding: Korean Health Technology R&D Project
(HI12C0014), Ministry of Health & Welfare, Republic of Korea.
Competing interest: None stated.
|
What is Already Known?
• The phenotype of Dent disease differs
across different regions of the world and there are no genotype
phenotype correlations.
• Progression to renal failure may occur in
third to fifth decades of life.
What This Study Adds?
• Dent disease has an early onset with severe
symptoms in this cohort of Indian children.
• Renal failure may occur in the first decade
of life with the most commonly described CLCN5 mutation
(Ser244Leu).
|
References
1. Dent CE, Friedman M. Hypercalciuric rickets
associated with renal tubular damage. Arch Dis Child. 1964;39:240-9.
2. Wrong OM, Norden AG, Feest TG. Dent’s disease: a
familial proximal renal tubular syndrome with low-molecular weight
proteinuria, hypercalciuria, nephrocalcinosis, metabolic bone disease,
progressive renal failure and a marked male predominance. QJM.
1994;87:473-93.
3. Scheinman SJ. X-linked hypercalciuric
nephrolithiasis: clinical syndromes and chloride channel mutations.
Kidney Int. 1998;53:3-17.
4. Pook MA, Wrong O, Wooding C, Norden AG, Feest TG,
Thakker RV. Dent’s disease, a renal Fanconi syndrome with
nephrocalcinosis and kidney stones, is associated with a micro-deletion
involving DXS255 and maps to Xp11.22. Hum Mol Genet. 1993;2:2129-34.
5. Hoopes RR Jr, Raja KM, Koich A, Hueber P, Reid R,
Knohl SJ, et al. Evidence of genetic heterogeneity in Dent’s
disease. Kidney Int. 2004;65:1615-20.
6. Hoopes RR Jr, Shrimpton AE, Knohl SJ, Hueber P,
Hoppe B, Matyus J, et al. Dent disease with mutations in OCRL1.
Am J Hum Genet. 2005;76:260-7.
7. Shrimpton AE, Hoopes RR Jr, Knohl SJ, Hueber P,
Reed AA, Christie PT, et al. OCRL1 mutations in Dent 2
patients suggest a mechanism for phenotypic variability. Nephron
Physiol. 2009;112:27-36.
8. Ludwig M, Utsch B, Monnens LAH. Recent advances in
understanding the clinical and genetic heterogeneity of Dent’s disease.
Nephrol Dial Transplant. 2006;21:2708-17.
9. Sethi SK, Ludwig M, Kabra M, Hari P, Bagga A.
Vitamin A responsive night blindness in Dent’s disease. Pediatr Nephrol.
2009;24:1765-70.
10. Hari P, Biswas B, Pandey R, Kalaivani M, Kumar R,
Bagga A. Updated height and creatinine based equation and its validation
for estimation of glomerular filtration rate in children from developing
countries. Clin Exp Nephrol. 2012;16:697-705.
11. Ludwig M, Doroszewicz J, Seyberth HW, Bökenkamp
A, Balluch B, Nuutinen M, et al. Functional evaluation of Dent’s
disease-causing mutations: implications for ClC-5 channel trafficking
and internalization. Hum Genet. 2005;117:228-37.
12. Lloyd SE, Gunther W, Pearce SHS, Thomson A,
Bianchi ML, Bosio M, et al. Characterization of renal chloride
channel, CNCN5, mutations in hypercalciuric nephrolithiasis
(kidney stones) disorders. Human Mol Genet. 1997;6:1233-9.
13. Takemura T, Hino S, Ikeda M, Okada M, Igarashi
T, Inatomi J, et al. Identification of two novel mutations in
the CLCN5 gene in Japanese patients with familial idiopathic low
molecular weight proteinuria (Japanese Dent’s disease). Am J Kidney Dis.
2001;37:138-43.
14. Wu F, Roche P, Christie PT, Loh NY, Reed AAC,
Esunof RM, et al. Modeling study of human renal chloride channel
(hCLC-5) mutations suggests a structural functional relationship. Kidney
Int. 2003;63:1426-32.
15. Dutzler R, Campbell EB, Cadene M, Chait BT,
MacKinnon R. X-ray structure of a ClC chloride channel at 3.0Å
reveals the molecular basis of anion selectivity. Nature.
2002;415:287-94.
16. Mansour-Hendili L, Blanchard A, Pottier NL,
Roncelin I, Lourdel S, Treard C, et al. Mutation update of the
CLCN5 gene responsible for Dent disease 1. Hum Mutat. 2015; 36:743-52.
17. Sekine T, Komoda F, Miura K, Takita J, Shimadzu
M, Matsuyama T, et al. Japanese Dent disease has a wider clinical
spectrum than Dent disease in Europe/USA: genetic and clinical studies
of 86 unrelated patients with low-molecular-weight proteinuria. Nephrol
Dial Transplant. 2014;29:376-84.
18. Anglani F, D’Angelo A, Bertizzolo LM, Tosetto E,
Ceol M, Cremasco D, et al., on behalf of Dent disease Italian
network. Nephrolithiasis, kidney failure and bone disorders in Dent
disease patients with and without CLCN5 mutations. Springer Plus.
2015;4:492-8.
19. Annigeri RA, Rajagopalan R. Hypophosphatemic
rickets due to Dent’s disease: A case report and review of literature.
Indian J Nephrol. 2009;19:163-6.
20. Bolino A, Devoto M, Enia G, Zoccali C,
Weissenbach J, Romeo G. Genetic mapping in the Xp11.2 region of a new
form of X-linked hypophosphatemic rickets. Eur J Hum Genet.
1993;14:269-9.
21. Kelleher CL, Buckalew VM, Frederickson ED, Rhodes
DJ, Conner DA, Seidman JG, et al. CLCN5 mutation Ser244Leu is
associated with X-linked renal failure without X-linked recessive
hypophosphatemic rickets. Kidney Int. 1998;53:31-7.
22. Bockenhauer D, Bichet DG. Inherited secondary
nephrogenic diabetes insipidus: concentrating on humans. Am J Physiol
Renal Physiol 2013;304:F1037-42.
23. Bustamante M, Hasler U, Leroy V, de Seigneux S,
Dimitrov M, Mordasini D, et al. Calcium-sensing receptor
attenuates AVP-induced aquaporin-2 expression via a calmodulin-dependent
mechanism. J Am Soc Nephrol. 2008;19:109-16.
24. Devuyst O, Thakker RV. Dent disease. Orphanet J
Rare Dis. 2000;5:28-35.
25. Becker-Cohen R, Rinat C, Ben-Shalom E, Feinstein
S, Ivgi H, Frishberg Y. Vitamin A deficiency associated with urinary
retinol binding protein wasting in Dent’s disease. Pediatr Nephrol.
2012;27:1097-102.
|
|
|
 |
|
