|
|
|
Indian Pediatr 2014;51:
919-920 |
 |
Delayed Presentation of Rickets in a Child
with Labyrinthine Aplasia, Microtia and Microdontia (LAMM)
Syndrome
|
|
Ankur Singh, *Mustafa Tekin, *Michelle Falcone and
Seema Kapoor
From the Department of Pediatrics, Division of
Genetics, MAMC & Associated Lok Nayak Hospital, New Delhi, India; and
*John P Hussman Institute for Human Genetics, University of Miami,
Miller Scool of Medicine, Miami, FL, USA.
Correspondence to: Dr Seema Kapoor, M-439, Ground
Floor, Guruharkishan Nagar, Paschim Vihar, New Delhi, India. Email:
[email protected]
Received: July 18, 2014;
Initial review: July 28, 2014;
Initial review: August 26, 2014.
|
|
Background: Labyrinthine Aplasia, Microtia and Microdontia (LAMM)
syndrome is characterized by the complete absence of inner ear
structures (Michel aplasia), microtia and microdontia. Hypophosphatemic
rickets results from defects in the renal tubular reabsorption of
filtered phosphate. Case characteristics: 13-year-old Indian girl
presented with deafness since infancy and progressive wrist widening and
genu valgum for last one year. Observation: Homozygous novel
missense mutation in fibroblast growth factor 3. Message: LAMM
syndrome and hypophosphatemic rickets may be associated.
Keywords: Deafness, Fibroblast growth factor
receptor-3, Hypophosphatemic rickets.
|
|
C
ongenital deafness with labyrinthine aplasia
(also known as Michel aplasia), microtia, and microdontia (LAMM
syndrome, OMIM #610706) is characterized by profound bilateral
congenital deafness associated with inner ear anomalies (most often
bilateral complete labyrinthine aplasia), type I microtia (typically
bilateral), and microdontia. LAMM syndrome is an autosomal recessive
condition and has been found in individuals with either homozygous or
compound heterozygous FGF3 mutations [1,2]. The FGF3 gene
encodes fibroblast growth factor 3, a protein that plays a critical role
in the embryonic development of the otic placode (which becomes the
inner ear) and its differentiation into the vestibular and cochlear
structures, the teeth, and external ears. Twelve FGF3 mutations
have been identified in individuals with LAMM syndrome including six
missense and six nonsense mutations or small deletions [2].
In this study, we report a 13-year-old girl with LAMM
syndrome and progressive hypophosphatemic rickets, which has not been
reported earlier, in association with LAMM syndrome.
Case Report
This 13-year-old girl was born to non-consanguineous
37-year-old mother and 37-year-old father. The pregnancy and birth were
uneventful. Deafness had been diagnosed in early infancy, and for the
past one year, she developed progressive genu valgum and wrist widening
and leg pain. Her temporary teeth had just started falling out for the
last few months. Except for delayed language, development in all three
domains was normal. There was no family history of deafness or skeletal
abnormalities. Anthropometry was within normal limits. She presented
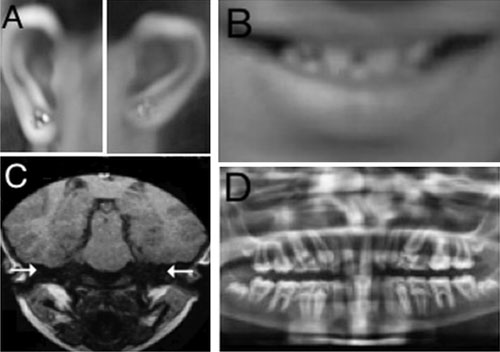
with type 1 microtia, widely spaced small teeth (Fig.1),
and genu valgum. Neurological examination was normal. On investigation,
she had profound sensorineural hearing loss; Magnetic resonance imaging
of her inner ear showed bilateral complete labyrinthine aplasia (Fig.1).
Orthopantogram revealed microdontia, generalized thinning of enamel, and
enlarged pulp (Fig.1). An X-ray of the patient’s
pelvis showed generalized osteopenia with an ill-defined pubis. X-ray
of knees showed fraying, cupping and splaying of metaphyses.
 |
|
Fig. 1 Clinical findings of the
proband: (a) Grade 1 microtia (b) Microdontia (c) Bilateral
complete labyrinthine aplasia (d) Microdontia, thin enamel, and
increased pulp.
|
Laboratory analyses of serum revealed following
values: inorganic phosphate 2.5 mg/dL, (Normal 3.5-6.6 mg/dL), calcium
8.5 mg/dL; alkaline phosphatase 1755 IU/L; intact-PTH, 55 pg/mL (Normal
9-65 pg/mL); 25-hydoxyvitamin D3, 28 ng/mL (Normal 10-53 ng/mL). Urinary
phosphate clearance was increased at 87 mL/min (normal 5–12 mL/min).
Sequencing analysis of the proband revealed a novel
homozygous change in FGF3 (c.534C>G) causing a phenylalanine to
leucine substitution at codon 178 (Web Fig. 1). Sequencing
analysis of the proband’s unaffected parents and sibling revealed that
they were all heterozygous for the FGF3 mutation. Analysis of
FGF23 in the proband showed no variation from the reference
sequence.
Discussion
The patient presented here had all three major
findings of LAMM syndrome, and was homozygous for an FGF3 mutation,
which confirms this diagnosis. Furthermore, the patient presented with
genu valgum; osteopenia with an ill-defined pubis; low serum phosphate
and elevated alkaline phosphatase despite supplementation with calcium,
phosphate, and vitamin D; and an elevated urinary phosphate clearance.
These physical findings and laboratory results were consistent with a
diagnosis of hypophosphatemic rickets.
Hypophosphatemic rickets is genetically hetero-geneous
condition and most commonly caused by mutation in PHEX gene, located on
X chromosome [3-6]. FGF23 mutation causes autosomal dominant form of
hypophosphatemic rickets [7]. We excluded the autosomal dominant form by
sequencing FGF23 gene. Possibility of X-linked form was unlikely as most
affected patients have early presentation incontrast to this patient who
presented with deformities at 13 years of age.
The phenylalanine residue at position 178 is highly
conserved among species from fish to primates (Web
Fig. 1). The two leucine amino acids located at both sides of
phe178 (leu177 and leu179) are implicated in the interaction of FGF3
with its cognate receptor [NCBI Reference Sequence: NG-009016.1]. Thus,
it is likely that an amino acid change in this region may alter the
receptor-ligand affinity and cause a loss of function, which is
confirmative for the pathogenesis in LAMM syndrome.FGF3 and FGF23 are
two members of the fibroblast growth factor (FGF) family. In humans, 22
members of the FGF family have been identified, all of which are
structurally related signaling molecules [8]. FGF family members possess
broad mitogenic and cell survival activities and are involved in a
variety of biological processes including embryonic development, cell
growth, morphogenesis, tissue repair, and tumor growth and invasion.
In absence of PHEX gene analysis, it is difficult to
put causative association between hypophosphatemic rickets and FGF3
mutation. The central role of FGF23 in its etiology and its structural
similarity to FGF3 lead us to hypothesize that the mutation found in
FGF3 could also be involved in the pathogenesis of hypophosphatemic
rickets in this patient. Our hypothesis needs to be substantiated with
functional assays on a larger cohort. At present, it seems a mere
association with a molecularly confirmed case of LAMM.
Acknowledgements: OscarDiaz-Horta and
Joseph Foster for their immense help in molecular work of this family.
Contributors: SK, AS: collected the data and
wrote the paper, which was edited and corrected by TM; MF: edited and
corrected the manuscript, helped in critical appraisal and performed the
molecular work.
Funding: None; Competing interests: None
stated.
Reference
1. Tekin M, Hismi BO, Fitoz S, Ozdag H, Cengiz FB,
Sirmaci A, et al. Homozygous mutations in fibroblast growth
factor 3 are associated with a new form of syndromic deafness
characterized by inner ear agenesis, microtia, and microdontia. Am J Hum
Genet. 2007;80:338-44.
2. Alizadeh Naderi AS, Reilly RF. Hereditary
disorders of renal phosphate wasting. Nat Rev Nephrol. 2010;6:657-65.
3. Ordonez J, Tekin M. Congenital Deafness with
Labyrinthine Aplasia, Microtia, and Microdontia. In: Pagon RA,
Adam MP, Bird TD, Dolan CR, Fong CT, Stephens K, editors. GeneReviews™
[Internet]. Seattle (WA): University of Washington, Seattle; 1993-2014.
Available from http://www.ncbi.nlm.nih.gov/books/NBK100664.
Accessed May 14, 2014.
4. Jan de Beur SM, Levine MA. Molecular pathogenesis
of hypophosphatemic rickets. J Clin Endocrinol Metab. 2002;87:2467-73.
5. Cho HY, Lee BH, Kang JH, Ha IS, Cheong HI, Choi Y.
A clinical and molecular genetic study of hypophosphatemic rickets in
children. Pediatr Res. 2005;58:329-33.
6. Bielesz B, Klaushofer K, Oberbauer R. Renal
phosphate loss in hereditary and acquired disorders of bone
mineralization.Bone. 2004;35:1229-39.
7. Econs MJ, McEnery PT, Lennon F, Speer MC.
Autosomal dominant hypophosphatemic rickets is linked to chromosome
12p13. J Clin Invest. 1997;100:2653-7.
8. Krejci P, Prochazkova J, Bryja V, Kozubik A,
Wilcox WR. Molecular pathology of the fibroblast growth factor family.
Hum Mutat. 2009;30:1245-55.
|
|
|
 |
|
