|
|
|
Indian Pediatr 2012;49: 924-925
|
 |
Early Presentation of Neuromyelitis Optica
|
|
Nikola Dimitrijevic, Dragana Bogicevic, *Aleksandar
Dimitrijevic and Dimitrije Nikolic
From the Department of Neurology; and *Educational
Department, University Children’s Hospital, and Medical Faculty
University of Belgrade, 11000 Belgrade, Serbia.
Correspondence to: Dr Nikola Dimitrijevic, University
Children’s Hospital, Neurology Department,
Tirsova 10, 11000 Belgrade, Serbia.
Email:
ndimitrijevic@beotel.net
Received: February 22, 2012;
Initial review: March 15, 2012;
Accepted: July 06, 2012.
|
Neuromyelitis optica is a rare autoimmune demyelinating disease of the
central nervous system in childhood. Its relapsing form is usually
reported in adults. We report a 3-year-old girl with relapsing, IgG
seropositive neuromyelitis optica. Initially she presented with optic
neuritis, followed by three relapses with deterioration of optic
neuritis and developing transverse myelitis. With each relapse, the
treatment was less effective. Four years after the onset of the disease,
the patient was blind, had paraplegia associated with urinary and bowel
incontinence and short stature.
Key words: Childhood, Neuromyelitis optica, Relapsing form.
|
|
Neuromyelitis optica is an inflammatory
demyelinating disease of the central nervous system clinicaly
presenting as optic neuritis and transverse myelitis [1,2]. The
onset ranges from early childhood to late adulthood with the mean
age in the forties. Pediatric case series data are insufficient,
especially for patients younger than six years [3,4].
Case Report
A previously healthy 3-year-old girl presented
with sudden visual loss preceded by pain while moving the left eye.
No recent history of fever, respiratory or gastro-intestinal
symptoms, infections, trauma, or vaccination was reported. The
family history was negative for neurological and autoimmune
disorders. Neurological evaluation revealed divergent strabismus,
mydriasis, and diminished pupillary reflex of the left eye.
Fundoscopy showed left optic disc pallor. Visual evoked potentials
were absent on the left eye, and significantly prolonged with
diminished amplitudes on the right eye. Brain MRI revealed slight
enlargement of the optic nerves, pre-dominantly on the left side.
Complete blood count and biochemical testing was normal. Analysis of
the cerebrospinal fluid showed normal findings with absent
oligoclonal bands. Serological tests for Toxoplasma, Borrellia
burgdorferi, Mycoplasma pneumoniae, Herpes simplex virus,
Epstein-Bar virus, cytomegalovirus, adenoviruses, influenza A and B
viruses, varicella zoster virus and HIV were negative. Antinuclear,
antiphos-pholipid and anti-thyroid antibodies, LE cells and
rheumatoid factor were negative. Levels of complement were within
normal ranges. The patient was treated with intravenous
methylprednisolone 20 mg/kg/day for 5 consecutive days, followed by
oral prednisone. Clinical improvement was noticed 2 weeks later with
visual function recovery on visual evoked potentials (complete on
the right and partial on the left eye).
The first relapse occurred 17 months later. The
patient developed flaccid paraparesis with increased tendon reflexes
and loss of sphincter control, preceded by back pain. This was
followed by bilateral loss of vision in the next five days. Brain
MRI was normal. Spine MRI showed a signal increase in spinal cord
between T8 and T12 levels on T2–weighted images. Intravenous
methylprednisolone therapy was repeated. Ten days after the
initiation of the steroid therapy the patient regained vision of the
right eye, with improved muscle strength and nearly normal muscle
tone of lower extremities. A month later sphincter control was
established. Spinal cord lesion on the control MRI disappeared
completely.
 |
|
(a)
(b)
(c)
|
|
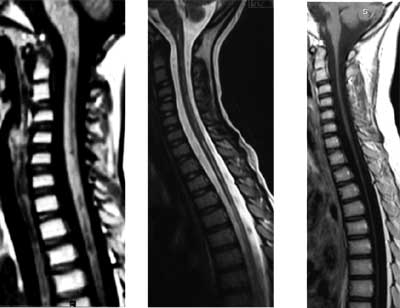
Fig. 1 Progression of spinal cord
atrophy shown on MRI (a) at the second relapse; (b) at the
third relapse; and (c) after third relapse.
|
Second relapse occurred at the age of 5 years and
10 months, with loss of vision in the right eye and weakness in the
arms and legs. There was moderate hypertonia in legs with Babinski
sign and increased tendon reflexes. Visual evoked potentials showed
no cortical responses. Brain MRI was normal. Spine MRI revealed
enhanced signal on T2 weighted images between C1 and T2 levels (Fig.
1a). Intravenous methylprednisolone was administered, with
significant improvement after two weeks. Patient was able to walk,
climb the stairs and jump. Muscle tone of the arms and legs was
normalized, but the left eye blindness persisted.
The third relapse occurred at the age of 6 years
and 4 months with total loss of vision and inability to sit, stand
and walk. Vibratory sensory loss was noted below T10 level. Brain
MRI showed atrophy of both optic nerves, optic chiasm and optic
tracts . Spine MRI revealed diffuse cervical and thoracic spinal
cord atrophy with subsequent dilatation of the central canal (hydromyelia)
(Fig. 1b). Intravenous methylprednisolone and
immunoglobulin treatement was unsuccessful. Parents of the patient
refused any further therapy.
An year later, neuromyelitis optica testing
revealed high titer of antibodies.
At the age of nine years the patient was blind
with spastic paraparesis, sensory loss below T10 level, urinary and
bowel incontinence, neurogenic bladder with recurrent urinary
infections, and height below third percentile. Mental development
was normal. MRI revealed severe atrophy of the spinal cord (Fig.
1c).
Discussion
Relapsing neuromyelitis optica is rare in
children. The youngest case previously reported in literature was a
boy of 23 months [3]. Our patient presented with isolated optic
neuritis at the age of 3 years, transverse myelitis occurred 17
months later. At the time of the first relapse, our patient
fulfilled all absolute criteria, two out of three major supportive
criteria and all minor supportive criteria for the diagnosis of
neuromyelitis optica. NMO-IgG antibodies were confirmed later [5-7].
The diagnosis of neuromyelitis optica has been
considerably facilitated by the discovery of highly specific serum
autoantibody biomarker - NMO-IgG [6]. The high titer of NMO-IgG
correlates well with frequent relapses [8]. Patients with the
relapsing form of NMO have demonstrated progressive disability.
There are no scientifically proven guidelines and
treatment strategies either in the acute attacks or on a long-term
base. The outcome depends on early effective immunosuppression prior
to accumulation of severe neurological damage.
Our patient is one of the youngest cases with
relapsing NMO and NMO-IgG seropositivity reported in the literature.
Although rare, pediatric NMO needs specific attention due to an
unpredictable clinical course and possible poor outcome with severe
disability . It is very important to identify risk factors that
predict a relapsing course and to determine optimal treatment,
especially for long-term preventive immunotherapy.
Acknowledgments: Professor Robert Marignier
and Laboratory in Bron (France) for performing NMO-IgG analysis.
Professor Borivoj Marjanovic from Insitute for Mother and Child,
Belgrade, for useful clinical suggestions.
References
1. Wingerchuk DM, Weinshenker BG. Neuromyelitis
optica. Curr Treatment Options Neurol. 2008;10:55-66.
2. Weinshenker BG. Neuromyelitis optica is a
distinct from multiple sclerosis. Arch Neurol. 2007;64:899-901.
3. Yuksel D, Senbil N, Yilmaz D, Yavuz Gurer YK.
Devic’s neuromyelitis optica in an infant. J Child Neurol.
2007;22:1143.
4. Loma IP, Asato MR, Filipink RA, Alper G.
Neuromyelitis optica in young child with positive serum
autoantibody. Pediatr Neurol. 2008;39:209-12.
5. Wingerchuk DM, Lennon VA, Pittock SJ,
Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for
neuromyelitis optica. Neurology. 2006;66: 1485-9.
6. Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock
SJ, Lucchinetti CF, Fujihara K, et al. A serum autoantibody
marker of neuromyelitis optica: distinction from multiple sclerosis.
Lancet. 2004;364:2106-12.
7. Krupp LB, Banwell B, Tenembaum S. Consensus
definitions proposed for pediatric multiple sclerosis and related
disorders. Neurology. 2007;68:S7-12.
8. Banwell B, Tenembaum S, Lennon VA, Ursell E,
Kennedy J, Bar-Or A, et al. Neuromyelitis optica-IgG in
childhood inflammatory demyelinating CNS disordes. Neurology.
2008;70:344-52.
|
|
|
 |
|
