|
|
Case Reports Indian Pediatrics 2006;43:998-1000 |
||||
|
Laugier-Hunziker Syndrome |
||||
|
Kabir Sardana From the Departments of Pediatrics*, and Dermatology, Maulana Azad Medical College, New Delhi 110 002 and associated *Chacha Nehru Bal Chikitsalaya, Geeta Colony, Delhi 110031, India.
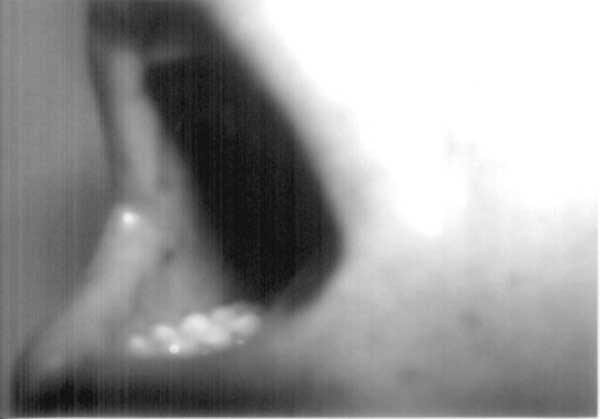
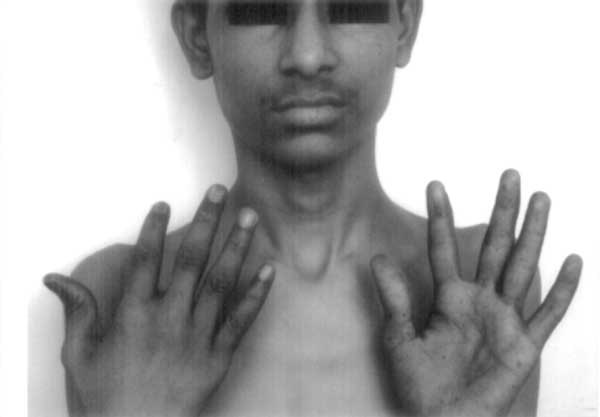
Peutz-Jeghers syndrome (PJS) is an autosomal dominant disorder characterized by melanocytic macules, multiple gastro-intestinal hamartomatous polyps, and an increased risk of various neoplasms(1). Diagnosis of this syndrome has life-long implications due to the high risk of malignancy and attendant life-long screening protocols. Laugier-Hunziker syndrome (LHS), on the other hand, is an acquired benign disorder having similar dermatological picture but no systemic manifestations(2). We herein describe a child with classical LHS to underline the importance of this differential diagnosis. The patient also had longitudinal melanonychia, a hitherto unreported finding in childhood. Case Report A 12-year-old male child with perioral, intraoral and palmar pigmentation since four years was referred for pediatric opinion by the dermatologist in view of complaints of poor weight gain and weakness. The child had a thin built and age-appropriate weight and height. There was no history of recurrent abdominal pain or vomiting, rectal bleeding, and no history suggestive of anemia. There was no drug or exposure history to explain the findings. The hair and uninvolved skin were normal. There was no suggestive family history with respect to dermatological or gastrointestinal manifestations of PJS; and both the parents and two other sibs (a 6-year- old male and a 14-year-old female) were without any suggestive symptoms. He was seen eight months back by a pediatric gastroenterologist with a probable diagnosis of PJS. Subsequently, he underwent one esophagogastroduodenoscopy with biopsy, one colonoscopy, and barium meal follow-up study for intestinal polyposis. These studies were normal. Blood pressure, blood biochemistry and hematology workup was within normal limits. The clinical diagnosis of PJS was reviewed in view of the lack of a family history of PJS, no gastrointestinal symptoms, and absence of polyps on endoscopic and imaging studies. On examination he was found to have bilateral, ill-defined, bluish black spots irregularly distributed over the dorsal aspect of the tongue (Fig. 1) and the gingivae as well as dark brown to black longitudinal bands affecting many fingers and toenails (Fig. 2). A biopsy of one melanotic macule of the patient’s tongue showed basal epidermal hyperpigmentation with moderate acanthosis and pigmentary incontinence. No dermatological abnormalities were noted in the other family members. Based on these dermatological findings and the biopsy features, a diagnosis of LHS was made. The parents were informed about the entirely benign nature of the disease with no systemic manifestations.
Discussion LHS is characterized by the presence of a variable number of asymptomatic pigmented lenticular or linear mucocutaneous macules. The color of the lesions varies from slate to brown-black(2). They can be isolated or confluent. The mouth and lips are most commonly involved. The corners of the mouth, the gingivae, the tongue, the fingers, and the plantar aspect of the feet are less frequent locations(3-5). The nails are affected in approximately 60% of patients(6) although this has never been seen in children. Histologically, there is increased basal keratinocyte melanin without expansion of the melanocytic population and superficial pigmentary incontinence with dermal melanophages. A definitive or even presumptive diagnosis of PJS was not valid in our patient as there was neither a family history nor intestinal polyps. Several conditions must be considered in the differential diagnosis of nail and mucocutaneous pigmentary abnormalities such as McCune-Albright syndrome, LEOPARD syndrome, Addison Disease, LAMB syndrome, Gardener syndrome and Cronkhite-Canada syndrome, as also benign racial pigmentation. Nail changes may also result from administration of systemic agents, especially chemotherapy(7). These conditions were ruled out on the basis of clinical examination for the associated conditions. Even though, in the absence of genetic studies a ‘forme-fruste’ form of PJS cannot be excluded, the distinctive distribution of pigmentation, lack of polyposis and nail involvement was in favor of the Laugier-Hunziker syndrome. In conclusion, LHS still remains a diagnosis of exclusion and given the intrafamilial and interfamilial variability of PJS, pigmentary changes suggestive of PJS in any patient need to be interpreted with caution. Moreover, pediatricians need to be aware of this benign acquired disorder to avoid mislabeling the patient as PJS leading to unnecessary and invasive diagnostic screening protocols | ||||
|
References | ||||
|
|
![]()