|
|
Brief Reports Indian Pediatrics 2006;43:974-979 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Etiology and Clinical Profile of Ambiguous Genitalia - An Overview of 10 Years Experience |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Rajesh R. Joshi, Sudha Rao and Meena Desai
Work up of patients with ambiguous genitalia requires a co-ordinated approach by pediatrician, surgeon, radiologist, and a good lab set-up to arrive at a quick diagnosis. We present here the clinical and etiological profile in 109 patients presenting with genital ambiguity seen over 10 years. Subjects and Methods Records of patients with ambiguous genitalia referred to our pediatric endocrine service were analysed. Detailed history including age of presentation, sex of rearing, consanguinity, family history of similar illness were noted. A thorough clinical examination including presence of hyperpigmentation, hypertension, associated anomalies or dysmorphic features were recorded. The genital ambiguity inclusive of phallic length, palpable gonads, position of urethral opening or presence of urogenital sinus and labial or labioscrotal folds were recorded to describe the stage of virilisation(1). Laboratory investigations included biochemical parameters, renal profile when indicated, blood sugar and hormonal studies inclusive of gonadotropins, sex steroids (testosterone done by chemiluminiscence) and specific precursor metabolites like 17-hydroxyprogesterone (17-OHP done by RIA) in some cases. HCG stimulation test was carried out by giving 1000 units IM on alternate days for 3 doses to study the testosterone and dihydrotestosterone (DHT done by RIA) synthetic response of the gonad. A twofold or more increase in testosterone level was considered a good response and a T to DHT ratio of more than 20 was considered as suggestive of 5 alpha reductase deficiency(2). Karyotype was also advised. Pelvic and abdominal sonography helped to determine the presence of mullerian structures and ovaries and occasionally to locate the undescended or partially descended testis. More invasive studies such as genitogram, laproscopy, surgical exploration and gonadal biopsy were done when indicated to identify the internal structures and ascertain gonadal morphology. On the basis of clinical and investigative profiles these children could be divided into 4 etiological categories as shown in Table I. TABLE I Etiology of Ambiguous Genitalia in 109 Patients
*Peno/perineoscrotal hypospadiasis with descended gonads
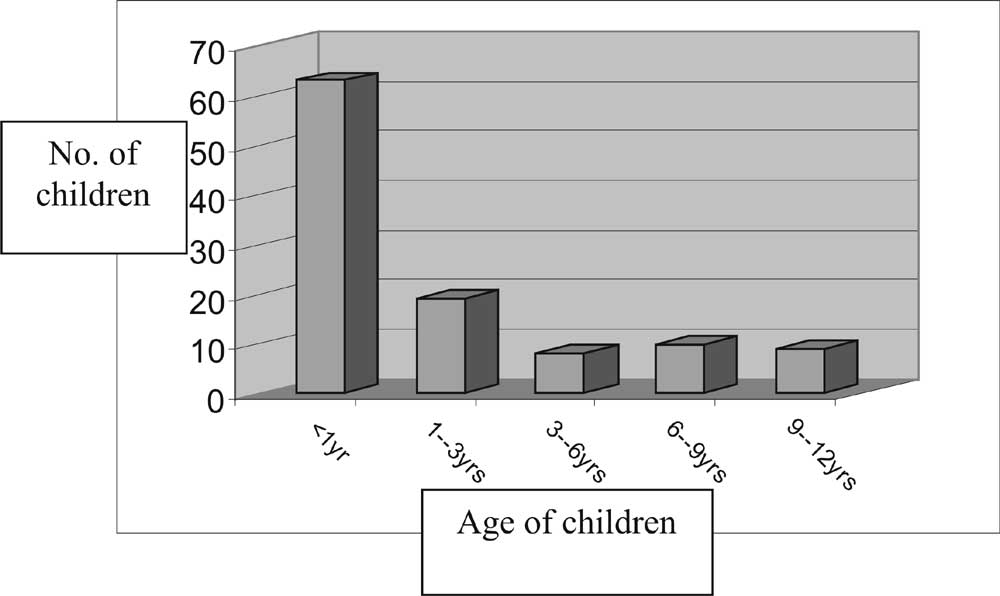
Results There was wide variation in the age of presentation of these 109 patients from 5 days to 12 years with a mean of 27.4 ± 38.4 months (Fig. 1). 63 patients (57.8%) presented prior to one year of age. History of consanguinity was elicited in 27 cases (24.7%). 65 patients (59.6%) were reared as males, 22 patients (20.2%) as females while 22 patients (20.2%) were not assigned sex of rearing at the time of presentation.
The various etiological groups are shown in Table I. All the patients presenting with FPH had congenital adrenal hyperplasia. Thirteen were born out of consanguinity. The diagnosis was based on presence of virilised external genitalia, nonpalpable gonads, presence of mullerian structures on imaging and confirmed by a elevated serum 17-OHP level (>20 ng/mL). 23 out of 30 patients (76.7%) had the salt wasting form(age of presentation 0.9 ± 1.2 months) presenting with hyponatremia and hyperkalemia while the remaining 7 (23.3%) were simple virilisers presenting over a wide range of age group (2 months to 11 years). There was skin hyperpigmentation in 13, failure to thrive in 18 and vomiting/diarrhea in 5 patients. Degree of virilisation ranged from Prader’s stage 1 (female external genitalia with clitoromegaly) to stage 4 (complete scrotal fusion with urogenital opening at the base or on shaft of penis)(3). Five patients out of 7 simple virilisers already had development of pubic and axillary hair and advanced bone age while 2 patients had hypertension at the time of presentation. Two out of these five patients had central precocious purberty. MPH (n = 57; 52.3%) constituted a major etiological group. Androgen insensitivity syndrome with 46 XY karyotype was diagnosed in 16 patients (28%) on the basis of normal/elevated basal testosterone, good testosterone response to HCG injection and absence of mullerian structures on imaging. Nine of these patients had elevated LH. Partial androgen insensitivity was diagnosed in 11 patients presenting with varying degrees of undervirilisation (penoscrotal/perineal hypo-spadias, undescended testis with hypospadias) and in 5 children complete androgen insensitivity was diagnosed based on presentation of female phenotype and inguinal swelling(containing gonads). All these patients had normal T/DHT ratio(< 20). Twelve children with 5 a reductase deficiency presented with penoscrotal hypospadias with descended gonads while one child had unilateral undescended testis with hypospadias along with 46 XY karyotype. HCG stimulated values of T/DHT ratio was uniformly high (>20). Ten patients (17.5%) with penoscrotal/perineal hypospadias with descended gonads were found to have normal Leydig cell reserve as there was a good rise in testosterone after HCG stimulation. DHT levels could not be done in these cases. Diagnostic possibilities in these patients were androgen insensitivity syndrome or 5 a reductase deficiency. HCG unresponsiveness was found in 4 cases (7.1%). These patients had bilateral undescended testes without any mullerian structures on imaging. Further investigations in them confirmed of having intra-abdominal testes in 2 while the remaining 2 had anorchia(vanishing testes syndrome). There were 4 additional patients with HCG responsiveness, no mullerian structures and bilateral undescended testes which were located at either inguinal or abdominal sites. One child with MPH had persistent mullerian duct syndrome presenting with unilateral inguinal hernia containing uterus and fallopian tubes and unilateral cryptorchidism. In 9 patients presenting with penoscrotal/perineal hypospadias with descended testes investigations could not be completed. Among the 9 patients with gonadal dysgenesis two patients had pure gonadal dysgenesis. They had external ambiguity in form of poorly formed phallus (<2 cm), single perineal opening and poorly formed scrotum. Laproscopy revealed hypoplastic fallopian tubes and uterus with bilateral streak gonads which were confirmed on biopsy. Other 7 patients were of mixed gonadal dysgenesis. They had small phallus and single streak gonad. Four of them had hypoplastic uterus while 3 patients had hypoplastic vas with unicornuate uterus. One patient had features of Aarskog syndrome whereas other child presenting with nephrotic syndrome had Denny Drash syndrome. Nine patients could not be categorized into any group due to incomplete investigations. Discussion It is heartening to note that most patients presented to us below one year (57.8%) contrary to a study from North India where 70% patients presented after 5 years(4). Other study from South India showed 60% patients presenting by 2 years(5) quite similar to ours. CAH is the commonest etiological factor causing ambiguous genitalia(5-8) as found in our case series (27.5%). One report(6) had as high as 72% of patients with CAH in their case series of ambiguous genitalia which ascertains the need to exclude CAH and avert a salt losing crisis in genetic females presenting with virilisation(1). Over 90% of CAH have the 21-hydroxylase deficiency, with built up of 17-OHP, a by-product prior to the block(7) as seen in our patients. Two patients having hypertension were suspected of having 11-b hydroxylase deficiency, however deoxy-corticosterone level which is a precursor by-product was not available for confirmation. In our series, sixteen cases of CAH were diagnosed in the newborn period. One study(4) emphasizes the need for early diagnosis and proper treatment to prevent morbidity like hirsuitism, premature pubarche and short stature as seen in few of our patients. MPH is the most common group and accounts for more than 50% cases of intersex disorders(7) as seen in our study. Androgen insensitivity syndrome and 5-a reductase deficiency were common causes of MPH in our series. Other studies have also reported large number of cases with AIS i.e., 90% and 88% of patients(9,10) and AIS/5-a reductase deficiency in 46.8% patients(8). Though increased levels of LH are seen in AIS(11), as present in 9 of our patients, it is not a universal finding(12). It is important to consider CAIS in any female patient with inguinal swelling(13). Partial AIS presents with wide variety of phenotypic conditions(14) as seen in our patients. 5-a reductase deficiency can present with different phenotypes according to severity of masculinization defects(15). Our patients presented with type 2 and 3 phenotypes(15). Differentiating this group from androgen insensitivity is important as patients with 5-a reductase deficiency masculinize at puberty presumably due to direct action of testosterone on the phallus(16). HCG stimulation test is an important work up to differentiate between normal Leydig cell reserve i.e., good response to HCG (seen in 14 of our patients) and HCG unresponsiveness (4 patients). Patients with HCG responsiveness have AIS or 5-a reductase deficiency while those with unresponsiveness could be testosterone biosynthetic defects, vanishing testes, Leydig cell agenesis, Leydig cell hypoplasia, abnormal Leydig cell gonadotropin receptors or delayed receptor maturation. Very few centers offer endocrine assays to identify the buildup of precursor products in condition of testosterone biosynthetic defects and receptor studies are not available easily. Reaching etiological diagnosis in cases of MPH is difficult because of variability of individual cases(9). The causes of MPH are also numerous and heterogenous(17,18). Detailed diagnostic work up like genital skin biopsy for androgen receptor binding assay and analysis of 5-a reductase activity is not available easily. Etiology may not be established as the exhaustive diagnostic workup may not be completed(17) as seen in 9 of our patients. Disorders of gonadal differentiation requires more invasive investigations. Confirmatory diagnosis of true hemaphroditism was by finding bilateral ovotestes on open biopsy of the gonad. This is the rarest variant of intersex disorders with 46,XX as the commonest karyotype in more than 60% patients(2,19). Both of our patients had 46,XX karyotype. Gonadal dysgenesis patients usually have karyotype 46,XY (8 patients) or 45,X/46,XY (2,20). To conclude pediatricians have a key role in relieving psychological distress of families and patients by co-ordinating the diagnostic evaluation , helping families understand their child’s medical condition and maintaining open communication between family and other health care team. Acknowledgement We are grateful to Dr. Dod, Chief Senior Executive of B.J. Wadia Hospital for Children for giving permission to publish this article. Contributors: RRJ reviewed and analyzed the data and drafted the manuscript. SR helped in analyzing data and preparing the manuscript. MD was involved in the management of patients, revising the article critically and final draft of the article. Competing Interests: None. Funding: Nil.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
References | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
![]()