|
|
Review Article Indian Pediatrics 2004;41:1115-1123 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Koumudi Godbole
Abstract Hirschsprung’s disease (HSCR) is the main genetic cause of functional intestinal obstruction with an incidence of 1/5000 live births. The etiology of HSCR is complex and is presumed to be a sex-influenced multifactorial disorder, with contributions from several genes. All the genes involved in HSCR are also involved with the early development of the enteric nervous system. HSCR is known to be associated with a chromosomal abnormality in 12% of cases, and with other congenital anomalies in additional 18% of cases. It is recommended that patients, including newborns, with HSCR undergo a careful assessment by a clinician trained in dysmorphology. Echocardiography, ultrasonography for urogenital malformations and skeletal x-rays should be routinely performed in cases with HSCR to rule out associated anomalies. HSCR associated with dysmorphic features or any additional systemic anomaly should prompt chromosomal studies. Genetic counseling should be provided to families of HSCR patients as the recurrence risk varies from 4% to up to 50% depending on whether it is non-syndromic or part of a specific syndrome. Key words: Genetic heterogeneity, Hirsch-sprung’s disease. Hirschsprung’s disease (HSCR, aganglionic megacolon) is the main genetic cause of functional intestinal obstruction with an incidence of 1/5000 live births. It was first described by Harald Hirschsprung in 1888, in two unrelated boys who died from chronic severe constipation with abdominal distension resulting in congenital megacolon. HSCR is of particular interest to Geneticists as it is known to be associated with a chromosomal abnormality in 12% of cases and with an additional 18% of cases with other congenital anomalies(1). The aim of this review is to discuss embryo-pathogenesis, clinical features and molecular findings in HSCR. Embryology and pathogenesis: Hscr as a neurocristopathy The neurocristopathies are a heterogeneous group of disorders resulting from impaired growth, defective differentiation and migration of the neural crest cells(2,3). The neural crest is a part of the folding neural tube that pinches off to form the cell bodies of all neurons and supporting cells outside the central nervous system(4). Neural crest cells follow specific migratory pathways to differentiate into cell types such as neurons and glia of the sensory, sympathetic and parasympathetic ganglia, neuroendocrine cells, adrenal medulla, ocular connective tissue, pigmented cells, and facial cartilage(5). HSCR is characterized by the absence of parasympathetic intrinsic ganglion cells in the submucosal and myenteric plexuses of the enteric nervous system, resulting from premature migration arrest of neural crest cells in the hindgut between 5 and 12 weeks of gestation. The vagus nerve and pelvic splanchnic nerves cannot synapse within the plexus to effect contraction of the colon and, therefore feces accumulate(3,6). Dilatation of the colon results from failure of peristalsis in the distal aganglionic segment, which prevents movement of the intestinal contents(7). HSCR is further classified into ultra short segment, short segment, long segment, total colonic, and total intestinal types depending on the length of the aganglionic segment, with the internal anal sphincter as the constant inferior limit(1). HSCR and autonomic dysfunction Recent literature suggests that a more widespread autonomic dysfunction may be associated with apparently isolated HSCR(8,9). Croaker, et al.(10) estimated that about 1.5% of all HSCR patients and 10% of those with total colonic aganglionosis may develop congenital central hypoventilation syndrome, and as many as 50% of patients with congenital central hypoventilation syndrome may also develop HSCR. Genetic epidemiology The incidence of HSCR varies significantly among ethnic groups: 1.5, 2.1, and 2.8 per 10,000 live births in Caucasians, African-Americans, and Asians, respectively(11). HSCR is isolated (non-syndromic) in 70% of cases, whereas it is associated with other congenital malformations (including the syndromic form) in 18% of cases and with a chromosomal abnormality in 12%(1). The congenital malformations commonly reported with HSCR are cardiac (5%), renal (4.4%), genital (2-3%). Isolated HSCR appears to be a multifactorial defect with sex modified penetrance and variable expression in the length of the aganglionic segment. Familial forms of HSCR represent 6-15% of the cases(12). There is a preponderance of affected males, and the male to female sex ratio is 4:1(13). Short segment HSCR is far more frequent than long segment HSCR (80% and 20% respectively)(14). (a) Chromosomal anomalies A chromosomal abnormality is found in 12% cases of HSCR, with Down syndrome (trisomy 21) being by far the most frequent. The chromosomal anomalies reported with HSCR are listed in Table I(19-21). Several frequently reported interstitial deletions in affected individuals led to the identification of HSCR predisposing genes, including RET at 10q11.2 (15, 16), EDNRB at 13q22.1(17), and SIP1 (ZFHX1B) gene at 2q22(18). TABLE I Chromosomal Anomalies Reported with HSCR.
Ref: 1, 15-21 (b) Syndromes and associated anomalies HSCR can occur as part of a recognized syndrome or in association with a wide range of other congenital anomalies. Table II summarizes syndromes commonly associated with HSCR. TABLE II Syndromes Reported with HSCR.
Neurocristopathies Shah-Waardenburg syndrome (MIM 277580) The combination of HSCR and Waardenburg syndrome is known as Shah-Waardenburg syndrome (WS4), a genetically heterogeneous condition that can be inherited in an autosomal recessive or autosomal dominant manner. In addition to HSCR, patients with Shah-Waardenburg Syndrome have pigmentary anomalies and sensorineural deafness. Homozygous mutations of the endothelin pathway and heterozygous SOX10 mutations have been identified in WS4 patients(22,23). Patients carrying a SOX10 mutation may also present with CNS involvement including seizures, ataxia and demyelinating peripheral and central neuropathies(24). Haddad syndrome (MIM 209880) This syndrome is characterized by congenital central hypoventilation (CCHS), and HSCR, representing 14-20% of CCHS patients. In these cases, long segment HSCR is more frequent and the sex ratio is equal, contrary to the male preponderance observed in isolated HSCR(25,26). Mutations in the RET and endothelin signaling pathways have been identified in two patients with Haddad syndrome(27). Multiple endocrine neoplasia type 2/ MEN 2 (MIM171400) Multiple endocrine neoplasia type 2A (MEN 2A) is a familial cancer syndrome with autosomal dominant inheritance and high penetrance. It is characterized by bilateral and multicentric medullary thyroid carcinoma (MTC) arising from the calcitonin-secreting thyroid C cells, pheochromocytoma, and less frequently hyperplasia of the parathyroid glands. Multiple endocrine neoplasia type 2B (MEN 2B) includes MTC, pheochromo-cytoma, oral neuromas, ganglion-euromatosis of the digestive tract, and skeletal abnormalities. The only malignancy seen in familial MTC is medullary thyroid carcinoma. Different allelic gain of function mutations in the RET proto-oncogene are responsible for these three cancer syndromes. The range of RET mutations including insertions, deletions, nonsense, missense and splicing mutations is suggestive of a loss of RET function in HSCR. Both FMTC and MEN2A can be associated with HSCR in some families. Non-neurocristopathies Goldberg-Shprintzen syndrome (MIM 235730) and Mowat-Wilson syndrome Goldberg-Shprintzen syndrome is probably an autosomal recessive; multiple congenital anomaly-mental retardation (MCA-MR) syndrome consisting of HSCR, cleft palate, microcephaly, hypotonia, short stature with or without facial dysmorphic features including hypertelorism, prominent nose, synophrys, thick eyelashes and sparse hair(28). Iris coloboma is a variable feature of this syndrome(29). The genetic etiology of this syndrome has not yet been elucidated. This syndrome should not be confused with Shprintzen-Goldberg syndrome with cranio-synostosis. Mowat-Wilson syndrome(30) shares many clinical features of Goldberg-Shprintzen syndrome and has a distinct facial phenotype, including a square-shaped face with a prominent but narrow triangular chin, broad nasal bridge, saddle nose, open mouth, full or everted lower lip, posteriorly rotated ears and large, uplifted ear lobes with a central depression (orechiette pasta/red blood corpuscle configuration). In contrast to Goldberg-Shprintzen syndrome, HSCR is not a mandatory feature of this syndrome. Congenital heart disease (45%), genitourinary anomalies including hypospadias (60%) and agenesis of corpus callosum (42%) are other common features associated with Mowat-Wilson syndrome. This is a sporadic syndrome resulting from de novo deletions or heterozygous mutations in ZFHX1B (SIP1). For genetic counseling purpose, Mowat-Wilson syndrome needs to be distinguished from the Goldberg-Shprintzen syndrome as the recurrence risk of the latter is up to 25%, whereas the former occurs sporadically. It has been suggested that for patients with HSCR with no mutations in SIP1, who have dysmorphic facial features, microcephaly and MR, the eponym Goldberg-Shprintzen syndrome could be kept(31). A different and yet unidentified gene must be responsible for this form of syndromic HSCR. The phenotypic overlap with the Mowat-Wilson syndrome suggests that it might be found in the SIP1 pathway(32). Cartilage-Hair Hypoplasia/CHH (MIM 250250) CHH is characterized by short limb dwarfism caused by metaphyseal dysplasia, fine, sparse blonde hair, transient microcytic anemia and immunodeficiency. It is caused by mutations in RMRP, a gene mapped to chromosome 9p13. HSCR is found in about 10% of cases of CHH(33). HSCR with limb anomalies About half a dozen syndromes are reported with limb anomalies including hypoplasia of distal phalanges and nails, both pre and post-axial polydactyly, and associated HSCR. These syndromes are listed in Table II (34-40). Molecular basis The etiology of non-syndromic/isolated HSCR is complex and is presumed to be multifactorial, with contributions from several genes and possibly environmental factors. All the genes involved in HSCR are also involved with the early development of the enteric nervous system. Loss-of-function mutations of RET have been identified in 50% of all familial and 15-35% of sporadic HSCR cases. Mutations in EDNRB, a component of endothelin signaling pathway account for about 5% of cases of HSCR. Mutations in seven other genes including those encoding RET ligands (GDNF and NTN), components of endothelin signaling pathway (EDN3, ECE-1), and the transcription factors SOX10, ZFHX1B and PMX2B (PHOX2B) have been occasionally identified in HSCR patients. L1CAM, a gene that encodes a neural cell adhesion molecule is suggested to have a modifying effect on one of the genes required for population of the gut by ganglion cell precursors(41). The genes involved in HSCR are listed in Table III. TABLE III Genes Involved in HSCR.
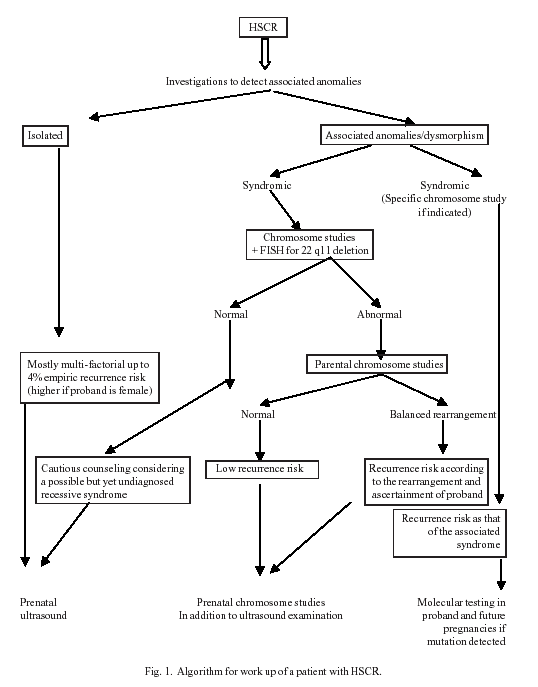
RET signaling pathway RET proto-oncogene was the first HSCR susceptibility gene identified and is expressed in the developing central and peripheral nervous system. Pathogenic mutations in RET occur throughout the gene and lead to misfolding or failure to transport the protein to the cell surface, resulting in haplo-insufficiency (half the gene dosage is not enough)(42). Mutations in GDNF and NTN, ligands for RET are described in patients with HSCR but are neither necessary nor sufficient to cause HSCR(43). Endothelin signaling pathway EDNRB, its ligand endothelin 3 (EDN3), and an associated enzyme, endothelin converting enzyme (ECE1) constitute the other major pathway in HSCR. Carrasquillo, et al.(44) proposed that an interaction between the RET and the ENDRB pathways at molecular and biochemical level may form the basis for HSCR. A murine model inter-crossing Ret null knockout mice with EDNRB hypomorphic piebald mice alleles confirmed the interaction of Ednrb and Ret in creating the HSCR phenotype. Arighi, et al.(45) have proposed an explanation as to how a single mutation in RET is associated with loss-and gain-of-function effects and can lead to opposing disease phenotypes. They analyzed the biological effects of classical loss-of-function (HSCR-associated) and gain-of-function (MEN2 causing) mutations in Ret, and compared these mutations with the double-faced "Janus" mutation at Cys620 of Ret. Like the two mutually incompatible faces of the Roman god Janus, this mutation causes both HSCR and MEN2 in a sizeable fraction of families. This raises the question whether all subjects with HSCR, regardless of a non-contributory family history, should be screened for RET exon 10 and 11 mutations to rule out cancer predisposition(1). For almost every HSCR gene, incomplete penetrance of the HSCR phenotype has been observed, probably due to genetic modifier loci. Thus, HSCR presents as a complex polygenic disorder with interplay of different genes. There is a clearly evident familial aggregation but Mendel’s laws do not seem to apply. In HSCR, oligogenic (involving a small number of genes) inheritance applies in most cases especially with short segment disease(14). Even within families with apparent monogenic inheritance of HSCR (usually long segment disease), the pheno-typic severity can be broad within members of the same family. Recommendations and conclusions 1. Patients including newborns with HSCR better require a careful assessment by a clinician trained in dysmorphology. 2. Cardiac and urogenital ultrasound and skeletal X-rays should be routinely performed in cases with HSCR to rule out associated anomalies. 3. HSCR associated with any additional systemic anomaly should prompt chromo-somal studies. 4. Prenatal detection of intestinal obstruction suggestive of HSCR, even in isolation, should prompt the clinician to offer inva-sive testing for fetal chromosomal studies. 5. Genetic counseling should be offered to the families of HSCR patients. HSCR is a sex influenced multifactorial congenital malformation with an overall recurrence risk of 4% in the sibs of proband; with a higher risk for a male sibling of a female proband. Recurrence risk for the syndromic variety varies according to the inheritance pattern of the syndrome. 6. Poor genotype-phenotype correlation in HSCR does not encourage routine muta-tional screening. However, RET mutation screen may be advisable owing to cancer predisposition. 6. An algorithm for workup of a patient with HSCR has been shown in Fig. 1.
Acknowledgement I gratefully acknowledge Dr. Marjan M. Nezarati, Clinical Geneticist and Program Director and Professor Dr. Ahmad S. Teebi, Section Head, Clinical Genetics, Division of Clinical and Metabolic Genetics, Hospital for Sick Children, Toronto, Canada for a critical review of the manuscript and valuable suggestions.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
![]()