The term muscular dystrophy encompasses a group of
inherited disorders characterized by progressive muscle weakness. The
best known are "X-linked dystrophinopathies", Duchenne muscular
dystrophy (DMD) and Becker muscular dystrophy (BMD)–a milder form which
together account for more than two-thirds of muscular dystrophy
patients. A clinically and genetically heterogeneous group presenting
with weakness of the pelvic and shoulder girdles is that of the
limb-girdle muscular dystrophies (LGMDs). Their mode of inheritance can
be autosomal dominant (LGMD 1A, 1B, 1C) or autosomal recessive (LGMD
2A-H)(1).
Advances in immunohistochemistry and genetics have
allowed a better understanding and precise molecular classification of
these disorders. Sarcoglycanopathies (SGPs) are autosomal recessive
LGMDs and are caused by mutations in any of the four sarcoglycan genes:
alpha (LGMD 2D), beta (LGMD 2E), gamma (LGMD 2C) and delta (LGMD 2F)(1).
We report a rare case of primary gamma-sarcoglycanopathy (SGP) which has
not been reported from India. This emphasizes the evolving new concept
in the field of muscular dystrophies" from dystrophinopathy to
sarcoglycanopathy".
Case Report
A nine-year-old boy born out of a non-consanguineous
marriage presented with history of walking on toes and broad based gait
noticed for the last two years. He belongs to Khatri community and is
from Budayun in Uttar Pradesh. There was associated difficulty in
running, climbing stairs, and getting up from the floor for the past 18
months. Over the last six months he started having frequent falls and
the parents noticed enlargement of muscles of the calf. All these
symptoms were progressively increasing. There was no associated muscle
pain, weakness of muscles of the arms, neck, back or face or difficulty
in chewing or swallowing. Birth history was non-contributory and
developmental miles-tones were normal. There was no family history of a
similar disease. He has a twin sister who is normal clinically and has
normal creatine phosphokinase levels.
On examination, the child had slightly elongated
facies. There was pseudohyper-trophy of both the calves, no contractures
and the child was able to walk unassisted but the gait was waddling and
toe based. Gower’s sign was positive. Muscle power was graded 3-4 for
the shoulder girdle and 2-3 for the hip girdle. Chest X-ray and
electrocardiogram were normal. The serum creatine phospho-kinase was
7330 IU/L (normal range: 15-195). Electromyography was consistent with a
myopathic pattern.
A biopsy of the left deltoid muscle was done. The
tissue was immediately transported and frozen in liquid nitrogen at
–160º C. 5 mm cryostat sections were prepared and stained for
histological examination. Hematoxylin and eosin (H&E) stained sections
showed variation in fiber size with central migration of nuclei.
Myophagocytosis was seen, with mild endomysial and perimysial fibrosis
and occasional regenerating fibers.
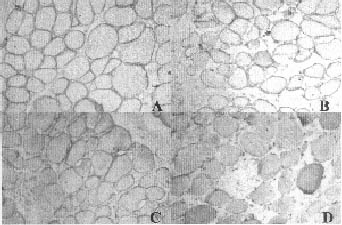
Some fibers were hypertrophied. Immunohistochemistry
(Fig. 1) for dystrophin I, II and III, alpha-sarcoglycan and
beta-sarcoglycan showed normal staining patterns but staining was
completely absent with antibodies to gamma-sarcoglycan.
 |
|
Fig. 1. Photograph showing positivity for
dystrophin (A, × 100), alpha-sarcoglycan (B, × 100), beta-sarcoglycan
(C, × 100) and negative staining for gamma-sarcoglycan (D, × 100). |
Gene deletion screening for 25 exons of the DMD gene
using multiplex PCR method identified no deletion. Thus a diagnosis of
primary gamma-sarcoglycanopathy was made. Mutations of sarcoglycan genes
could not be studied due to non availability.
Discussion
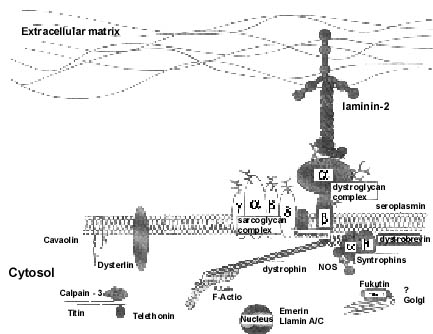
Soon after the discovery of dystrophin, the
dystrophin-associated proteins (DAPs), expressed on the sarcolemma, were
described (Fig. 2). Among them was the sarcoglycan (SG) complex,
a distinct group of five transmembrane proteins (alpha, beta, gamma,
delta and epsilon-sarcoglycans). This trans-membrane complex links the
cytoskeleton to the extracellular matrix and is essential for the
preservation of the integrity of the muscle cell membrane. Pathogenic
mutations in any of the SG genes (except epsilon-SG) disrupt the entire
SG complex and leads to secondary deficiency of the other SG
proteins(2).
 |
|
Fig. 2. Molecular architecture of dystrophin
and dystrophin associated proteins on the cell membrane. |
Mutations in the gamma-SG gene were first described
from North Africa. Tunisian patients with muscular dystrophy were
thought to have a defect in alpha-SG based on decreased immunostaining
with alpha-SG. However mapping to chromosome 13 ruled out a defect in
that gene and pointed to the gamma-SG gene at 13q12.
Primary SGP (including gamma-SGP) have been described
with a broad range of clinical presentations. Many cases have been
previously described under the name "SCARMD" (severe childhood autosomal
recessive muscular dystrophy) or DMD-like muscular dystrophy. However,
the term SCARMD used to describe the severity of the disease is not
always accurate as a description of the myopathy associated with primary
SGP because the phenotype may be milder, with juvenile or adult
onset(3). In fact, it has been reported that SGP caused by an identical
mutation in the gamma SG gene was characterized by either severe or mild
symptoms(4).
The diagnosis of SGP begins with documentation of
symptoms and signs, and elicitation of an accurate family history. They
have autosomal recessive inheritance, preferential and early involvement
of pelvic girdle muscles and subsequent involvement of shoulder girdle
muscles(5). Establishing autosomal recessive mode of inheritance may not
be easy but may be the only way of clinically distinguishing SGP from
dystro-phinopathy especially BMD(6). In SGP, there is absence of mental
retardation, facial and ocular muscles involvement and cardiac
involvement(5). Calvo, et al.(7) have however, reported right
ventricular hypertrophy and diastolic dysfunction among gamma SGP,
particularly in advanced stages of the disease. The clinical course of
gamma SGP is intermediate between DMD and BMD. They have onset of
weakness in childhood and become wheel chair bound by 25 years(5).
Immunohistochemical analysis is the most reliable
method of diagnosis. A mutation in anyone of the SG genes may lead to
secondary deficiency of other SG proteins, presumably due to
destabilization of the SG complex(10). The most frequently reported
pattern is that of multiple SG deficiencies in various combinations. In
a study on 25 patients, with SGP, Khadilkar, et al.(8) reported
84% with multiple SG deficiencies. Sarcoglycan gene mutation analysis is
essential to know the specific diagnosis in such cases with multiple
deficiencies. Though there may be equal loss of all the SG, generally
the one that stains most weakly is the one whose gene is mutated(2,9).
Gamma SGP patients may have normal or near normal alpha SG levels, hence
the screening with alpha SG can lead to underestimation of gamma SGP
cases.
Secondary dystrophin deficiency is well known in
patients with SG deficiency and is seen more often with gamma SGP(3). It
should also be remembered that the SG complex is remarkably reduced in
amount or even absent in DMD patients in addition to the absence of
dystrophin and gene defects are not detected in 30 percent of DMD
cases(10). When dystrophin appears at normal levels or is only slightly
decreased in amount, and no defect in the dystrophin gene is found, a
diagnosis of SGP is likely(6).
In conclusion, SGP is an example of muscular
dystrophy in which the same phenotype results from mutations in
different genes. The four muscular dystrophies belonging to this group
are indistinguishable not only clinically but also by
immuno-histochemistry (in case of multiple deficiencies), and molecular
studies are required to characterize the mutated gene. Accurate
diagnosis is important for prognosis, genetic counseling and possible
implications for gene therapy.
Contributors: VK and SG were incharge of the
case, SG and SL drafted the manuscript, VK critically reviewed the
manuscript and will act as guarantor for the paper, MCS reported the
muscle biopsy. All the authors were involved in finalization and
approval.
Funding: None.
Competing interests: None stated.
