|
|
Case Reports Indian Pediatrics 2002; 39:1050-1054 |
||
|
Partial 15q22 Trisomy due to Segregation of Maternal 10;15 Reciprocal Translocation |
||
|
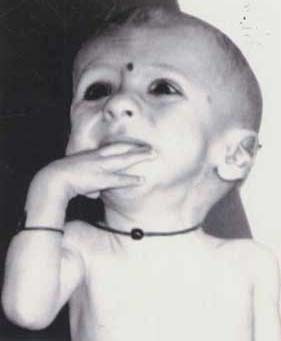
Structural chromosomal defects originating from a balanced translocation in either parents are often responsible for mental retardation and/or congenital malformations. The genetic counselling for balanced reciprocal translocation carriers is important in the management of future pregnancies(1). Analysis of duplication or deficiency of chromosomes in affected individuals can offer insight into the delineation of segmental aneuploidy syndromes. In the present report, we describe a case of partial trisomy 15q arising due to maternal reciprocal translocation involving chromosomes 10 and 15. Case Report An eight-month-old male child born to non-consanguineous parents was referred for genetic analysis with a history of develop-mental delay and failure to thrive. The father was 30 years old and the mother was 28 years old. The birth history was uneventful and the child had normal birth weight according to parents. The first pregnancy was an anencephalic female child who died after birth. The second pregnancy resulted in the birth of a female child who also died within the first month. The cause of death was not known except that the baby was very weak. The family history was non-contributing from father’s side and mother had a brother who was infertile and a sister with early cataract. On examination, the child had dysmorphic features in the form of hypertelorism, down slanting eyes, low-set large ears, prominent metopic suture, bulbous nose, mandibular hypoplasia and high arched palate (Fig. 1). Arachnodactyly, over riding of fingers and toes was noticed with hypoplastic creases. The child had short neck, deep sternal notch and scoliosis. The scrotal sac was underdeveloped with bilateral hypoplastic undescended testis. Neurologically the child was spastic with scissoring of legs and hypertonia. The child also had patent ductus arteriosus and lack of subcutaneous fat.
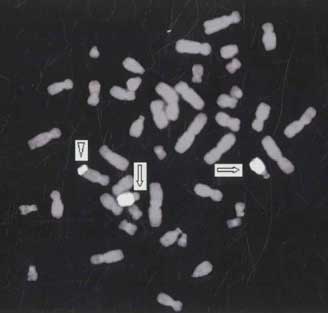
Metaphase chromosomes were prepared from peripheral blood lymphocyte cultures using standard cytogenetic protocols(2). Chromosomal analysis was done in GTG-banded metaphases(3). The child had 46 chromosomes, with an additional chromosomal segment on the long arm of chromosome 10. Father was cytogenetically normal and mother was a carrier of reciprocal translocation involving chromosomes 10 and 15. The translocation was observed in all metaphases analyzed. Fluorescence in-situ hybridization (FISH) was carried out using whole chromosome painting probes for chromosome 15 (VYSIS, USA). The denaturation, hybridization and signal detection were done according to the instruction manual supplied by the manufacturer. By combining G-banding and FISH results, the patient was found to have the karyotype 46, XY, der(10), t(10;15) (q26; q22) and mother was carrier of reciprocal translocation involving chromosomes 10 and 15 with chromosomal breakpoints q26 and q22 respectively and her karyotype was 46, XX, t(10;15) (q26;q22) (Fig. 2 and 3). The child had two normal 15 chromosomes and derived chromosome 10 with an extra segment of chromosome 15 from mother and normal chromosome 10 from father. Hence, the child was trisomic for 15q22 ® qter region.
Discussion After the description of 15q trisomy by Fugimoto et al(4), over 30 additional cases have been described with duplications of distal 15q trisomy, in which proximal breakpoints ranged from 15q21 to 15q26(5). In majority of patients, the 15q duplication was an unbalanced product of a parental reciprocal translocation with another autosome, most frequently another acrocentric chromosome(5,6). In two patients, the 15q trisomy was of post zygotic origin(7,8) and in one case the duplication was due to pericentric inversion. The present report is the first case of 15q22 trisomy due to reciprocal translocation between chromosomes 10 and 15 involving breakpoints q26 and q22 respectively, and also this is the first reported case of 15q trisomy from the Indian population. A specific phenotype is described in 15q trisomy with craniofacial, skeletal, neuro-logical and urogenital features(2). These include microcephaly, assymmetric face, downslanting and narrow palpebral fissures, bulbous nose, down-turned triangular mouth, long philtrum, high arched palate, low-set abnormal ears, short neck, scoliosis, structural abnormalities of fingers and toes, hypotonia and abnormal external genitalia. Commonly associated defects include seizures and congenital heart disease, a common cause of premature death in these children. However, in the present case hypertonia was observed instead of hypotonia and the child did not have seizures, which is a deviation from the main features of 15q trisomy. The carriers of balanced translocation are at about 9% risk of having children with unbalanced karyotype(9). Hence, prenatal cytogenetic analysis should be carried out in subsequent pregnancies in translocation carriers to offer best genetic counselling.
Acknowledgement Authors are thankful to Prof. Aubrey Milunsky and Dr. Herman Wyandt, Center for Human Genetics, Boston University for the help in FISH studies. This work was supported by a core grant to Center for DNA Fingerprinting & Diagnostics from the Department of Biotechnology, Government of India. Contributors: ARD and KP reviewed the literature and drafted the paper. ARD will act as guarantor for the paper. UD helped in the laboratory work and in drafting the paper. Funding: Core grant from the Department of Biotechnology, Government of India Competing interests: None stated.
| ||
|
References | ||
|
![]()