|
|
Case Reports Indian Pediatrics 2001; 38: 1298-1300 |
||
|
Massive Steatorrhea in a Child Due to Isolated Pancreatic Hypoplasia |
||
|
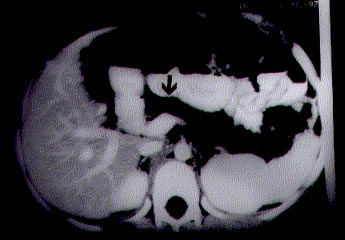
Steatorrhea due to developmental anomalies of pancreas is uncommon in children. Agenesis or hypoplasia of pancreas usually does not manifest with steatorrhea alone(1). We here report an unusual case who presented with massive steatorrhea and was noted to have pancreatic lipomatosis secondary to isolated pancreatic acinar hypoplasia. Case Report A 9-year-old boy presented with steatorrhea and failure to thrive since 1½ years of age. He was passing large volume, foul-smelling, greasy stools, 4-8 times per day. Frequency of motions used to increase with higher fat intake. Despite his preserved appetite he was not gaining weight and height. There was no history of pain abdomen, recurrent respiratory tract infection, poly-phagia, polyuria or polydipsia. The child was born of a non-consanguineous marriage. None of the family members had any pancreatic disorder or congenital malforma-tion. His height was 116 cm (88% of expected) and weight was 18 kg (70% of expected). There was no obvious congenital malformation. Rest of the systemic examina-tion was within normal limits. Hemoglobin, platelet counts, total and differential leukocyte counts were normal on several occasions done at an interval of 1 to 3 weeks. Skiagram of chest and abdomen did not reveal any abnormality. Liver and renal function tests, glucose tolerance test were within normal range. However, his fecal fat excretion was 26 g/day (normal <6 g/day). Five hours urinary D-xylose excretion and duodenal biopsy were also normal. Although his serum lipase and amylase levels were normal, fecal chymotrypsin and elastase levels were undetectable. His sweat chloride was 38 mmol per liter. The contrast enhanced C.T. scan of abdomen showed hypodense pancreas (Fig. 1) suggestive of pancreatic lipomatosis. There was no evidence of calci-fication. Endoscopic retrograde cholangio-pancreatography revealed a normal main pancreatic and bile ducts. With the above history and investigation findings, the diagnosis of pancreatic hypo-plasia with exocrine insufficiency was made. He was started on pancreatic enzyme replacement therapy, the dose of which was gradually increased till 5000 units of lipase per kg body weight per day. With this, diarrhea subsided and the child gained 6 kg of weight over 12 months time. Discussion The commonest cause of pancreatic insufficiency in children is cystic fibrosis. Other rare causes are Schwachman Diamond syndrome, Johanson Bizzard syndrome, Pearsons marrow pancreas syndrome, etc.(2-6). Lipomatous change of pancreas with normal pancreatogram has been reported in some of these conditions(2-4). However, they are associated with various other congenital malformations, skeletal changes and hematological abnormalities which were absent in our child. Children with isolated deficiency of lipase or colipase present with steatorrhea alone(2). Serum and duodenal lipase levels are very low or absent in them. Our child had normal serum lipase level and undetectable levels of elastase and chymotrypsin in feces. Various types of developmental anomalies of pancreas have been described but presentation with steatorrhea alone is rare. Complete agenesis is incompatible with prolonged survival(7). Agenesis of dorsal pancreas, ventral pancreas and uncinate process has been described and they are diagnosed on the basis of specific changes on pancreatogram and CT scan(1,7). Our patient had pancreatic lipomatosis with normal pancreatogram and normal blood sugar levels. This may be explained by pancreatic acinar hypoplasia(2). Such an occurrence has rarely been reported earlier(8). Lozano et al. described two such cases presenting with massive steatorrhea(8). Pancreatic hypoplasia has been reported more commonly as a part of various syndromes named earlier and in dia-betic patients(2-6). Treatment of pancreatic hypoplasia depends essentially on mode of presentation. Our child presented with steatorrhea only and he responded well to pancreatic enzyme replacement therapy. The long term outcome of such patients is not known because of rarity of the condition. Congenital pancreatic hypoplasia should always be kept in mind when a child manifests with gross steatorrhea.
Contributors: UP conceived and drafted the article. BRT helped in designing, drafting and also will act as the guarantor for the paper. SKS co-drafted the article. DKB helped in designing and drafting of the paper. Funding: None. Competing interests: None stated.
| ||
| References | ||
|
![]()