|
|
Case Reports Indian Pediatrics 2000;37: 1269-1274. |
|||||||||||||||||||||||||||||||||||||||||||||||||
|
Congenital Cystadenomatoid Malformation of Lung |
|||||||||||||||||||||||||||||||||||||||||||||||||
|
Congenital cystadenomatoid malformation (CCAM), a rare type of developmental anomaly of the lung was first acknowledged as a separate entity and introduced into English literature by Chin and Tang in 1949(1). Several patterns of clinical presentation have been observed: (i) still birth or neonatal death frequently associated with fetal hydrops, (ii) acute progressive respiratory distress in a newborn, (iii) an indolent course characterized by recurrent pulmonary infections, (iv) rarely pneumothorax(2), and (v) even more rarely as a prenatal diagnosis on maternal ultrasound or polyhydramnios(3). We report here four cases, a child with recurrent pulmonary infections, two neonates with respiratory distress one of whom had associated lung hypoplasia and cervical cord teratoma and a fetus with oligohydramnios associated with bilateral renal agenesis.
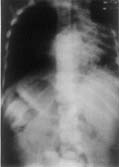
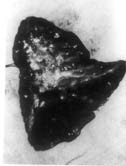
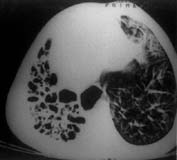
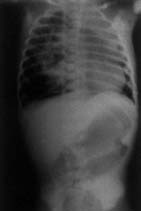
Case-1: A four-year-old boy was admitted with history of cough productive of yellowish, foul smelling sputum and fever off and on for 8 months. The child had severe growth failure, clubbing and halitosis. Right hemithorax was dull to percussion and had features of volume loss with crackles all over. Clinical impression was right sided bronchiectasis with collapse/fibrosis. Chest X-ray showed right sided hetero-genous opacity with ipsilateral mediastinal shift. Flexible fiberoptic bronchoscopy did not show any intra bronchial obstruction. Hemogram showed anemia with polymorpho-nuclear leukocytosis. Sputum and blood culture were non-contributory. Sweat electrolytes were within normal limits. He was treated with two weeks of parenteral antibiotics but continued to have cough off and on and was readmitted with similar clinical features. In addition he had drooping of the right shoulder. Chest X-ray showed diminished volume of right lung with multiple cystic shadows and rightward shift of mediastinum (Fig. 1). High resolution CT scan of chest showed the same findings, the entire right lung parenchyma had been replaced by cystic cavities 0.2-1 cm in size. Some showed fluid within them (Fig. 2). After treatment with antibiotics he was taken up for right pneumonectomy. Peroperatively the right lung was found to be riddled with cysts (Fig. 3). Histopathology of the resected lung showed congenital cystadenomatoid malformation type II. A section showing the lining epithelium is shown in Fig. 4.
The other three cases are listed in Table I.
Congenital cystic adenomatoid mal-formation (CCAM) of the lung is caused by anomalous fetal development of terminal respiratory structures, resulting in adenomatoid proliferation of bronchiolar elements and cyst formation leading to enlargement of the affected lobe. The clinical spectrum varies depending on the extent of malformation in the lung and associated conditions. Some authors have observed predilection of the right lung over the left for this anomaly(4). Involvement of an entire lung is distinctly rare as was seen in case 1. Single lobe involvement is the most common. Out of 153 cases reported till 1993 only 27 had multiple lobes affected(5). The other feature which was unique in the first case was ipsilateral mediastinal shift. Since the cysts are conglomerated into a well defined expansive mass, the space occupying lesion produces compression on surrounding tissues and pushes the mediastinum. This has been observed and reported in many cases earlier(6). A less common presentation is without mediastinal shift which is true about type II CCAM vis-a-vis type I(7). The probable reason for ipsilateral mediastinal shift in case 1 was fibrosis from repeated pulmonary infections. There was no associated pulmonary hypoplasia which is a well described associated anomaly(8). High frequency of other anomalies has been seen in type II CCAM(7). CCAM can be differentiated from other cystic lesions of the lung namely pulmonary sequestration, bronchogenic cyst, congenital lobar emphysema or diaphragmatic hernia and cystic bronchiectasis. In bronchiectasis, CT scan shows bronchi with thick walls sometimes looking like a string of pearls when aligned. In contrast, thin-walled cystic spaces are seen in CCAM(9). In the index case such subtle differentiation was not possible owing to severity of involvement. Histopathologically, a CCAM is charac-terized by varying sized cysts with polypoid configuration of the lining mucosa with absence of cartilage and inflammation. Mucous cells are absent in type II CCAM. In case 1, the cut section showed entire parenchyma replaced by numerous evenly spaced cysts less than 1 cm in diameter. Microscopically the cyst lining showed convolutions with cuboidal to tall columnar ciliated pseudostratified epithelium. The wall was composed of a layer of loose connective tissue with discontinuous bands of elastic tissue. There was nothing to suggest bronchiectasis. There was evidence of inflammation which may be present sometimes in CCAM where the lesion is silent until infection brings it to notice(9). The definitive treatment of CCAM is surgery. However, there is a controversy whether or not all cases be subjected to surgery. Surgery is indicated for the following reasons: (i) definite histological diagnosis, (ii) history of recurrent infections, and (iii) risk of malignancies which have been rarely reported(10). Our case 2 has been progressing uneventfully; however, a close follow up is being maintained. The outcome of lobectomy/pneumonec-tomy is good in children. In lobectomy the remaining lung grows and expands well enough so that total lung volume and pulmonary function tests return to normal(11). This response is most vigorous in the very young because new acini and alveoli form upto 5 years age(12). Post-resection majority of patients have an excellent result. Younger groups have lower ratio of residual volume to total lung capacity and higher maximum breathing capacity. This suggests that hyperplasia rather than overdistension occurs in the remaining lung(12). Prenatal diagnosis of cystic adenomatoid malformations by ultrasound has improved the management of the fetus as well helped to define the natural history and pathophysiology of this malformation(13). Although, the fetus being reported did not survive due to associated renal aplasia, antenatal diagnosis, maternal transport and immediate thoracotomy after birth can lead to survival. The association between CCAM and bilateral renal agenesis has been described before(14). The usual association of polyhydramnios with a fetus having CCAM has been ascribed to excessive lung fluid production by type II pneumocytes and by mechanical factors preventing fetal swallowing of lung fluid due to esophageal compression by the CCAM. However, association of oligohydramnios in Case 3 in the present series was due to bilateral renal agenesis. Association of cervical cord teratoma with CCAM has not been reported so far to the best of our knowledge. The parents of this child took a decision not to go in for surgery due to severe central nervous system defect. Although rare, recognition of CCAM pre- or post-natally is important to undertake surgery early and prevent the consequences of recurrent infections.
|
![]()