|
|
|
Indian Pediatr 2021;58:445-451 |
 |
Next-Generation
Sequencing for Congenital Nephrotic Syndrome: A Multi-Center
Cross-Sectional Study from India
|
|
Aditi Joshi,1* Aditi Sinha,2* Aakanksha Sharma,2 Uzma Shamim,1
Bharathram Uppilli,1 Pooja Sharma,1 Sana Zahra,1 Shaista Parveen,1
Aradhana Mathur,1 Monica Chandan,2 Prachi Tewari,2 Priyanka Khandelwal,2
Pankaj Hari,2 Mitali Mukerji,1 Mohammed Faruq,1 Arvind Bagga,2
for the NephQuest Consortium
From 1Genomics and Molecular Medicine, CSIR Institute of Genomics and
Integrative Biology, Delhi; and 2Division of Nephrology, Department of
Pediatrics, All India Institute of Medical Sciences, New Delhi; India.
*Joint first authors.
Correspondence to: Professor Arvind Bagga, Division of Nephrology,
Department of Pediatrics, All India Institute of Medical Sciences, New
Delhi, India.
Email: arvindbagga@hotmail.com
Dr Mohammed Faruq, Genomics and Molecular Medicine, CSIR Institute of
Genomics and Integrative Biology,
Delhi, India.
Email: faruq.mohd@igib.in
|
Objective: Information on etiology of congenital
nephrotic syndrome in non-Caucasian populations is limited. This study
aimed to determine the genetic basis of congenital nephrotic syndrome in
Indian patients. Methods: In this observational, cross-sectional
study, whole exome sequencing was performed on samples from all children
diagnosed with congenital nephrotic syndrome, presenting at centers
collaborating in a nationwide registry and biorepository. Analysis was
targeted to focus on reported or novel, pathogenic or likely pathogenic
variants in 89 genes implicated in etiology of nephrotic syndrome.
Sanger sequencing was used to confirm disease-causing variants in
patients and allelic segregation of compound heterozygous variants in
samples from parents. Inheritance of a shared haplotype was analyzed
among ten individuals carrying the most common variant. Results:
During 2017-2019, 34 patients with congenital nephrotic syndrome were
screened. Consanguinity and similar illness in siblings were reported in
eleven patients each. Homozygous or compound heterozygous, pathogenic or
likely pathogenic variants were found in NPHS1 in 24 cases,
including two novel variants. One patient each had homozygous pathogenic
or likely pathogenic known or novel variant in NPHS2, PLCE1,
OSGEP and LAMB2 genes. Patients with OSGEP and
LAMB2 mutations had phenotype typical of Galloway Mowat and Pierson
syndromes, respectively. Three variants in NPHS1 were common to
16 individuals. One reported variant in exon 19 (c.2600G>A; p.Gly867Asp)
appears to share a common founder. Conclusion: A genetic cause
was determined for 82.4% patients with congenital nephrotic syndrome.
Variants in NPHS1 are most common in Indian patients and founder
mutations
might be present.
Keywords: Nephrin, podocin, Galloway Mowat syndrome, Pierson
syndrome, NPHS1
|
|
C ongenital
nephrotic syndrome (NS) is a rare condition, characterized by
nephrotic range proteinuria, hypoalbuminemia and edema before 3
months of age. Most patients show morbidities related to edema,
infections and/or thrombosis, and progression to end stage renal
disease (ESRD) in early childhood [1]. An inherited basis is
reported in 60-80% patients; variants in NPHS1, which are
most frequent and also cause the Finnish type of congenital NS
[2], along with variants in NPHS2, PLCE1, LAMB2
and WT1, result in defects affecting proteins in the
podocyte slit diaphragm, actin cytoskeleton or transcription
regulation [3-5]. Existing reports on variants in Asian patients
are single-center and retrospective, screening for few genes
[6-10]. We describe here the results of next-generation
sequencing (NGS) in infants with congenital NS, enrolled
prospectively from April, 2017 to June, 2019, in a multicenter
collaboration on nephrotic syndrome.
METHODS
Following ethics approval and informed
parental consent, clinical details and blood samples were
collected from patients with congenital NS, diagnosed at seven
tertiary care centres in the country. Diagnosis required the
confirmation of nephrotic range proteinuria (spot urine protein
to creatinine ratio >2.0 mg/mg or dipstick 3+/4+ on three
occasions), hypoalbuminemia (serum albumin <3.0 g/dL) and edema
beginning below 3-months of age. Intrauterine infections and
structural renal anomalies were excluded by appropriate serology
and ultrasonography, respectively. In consonance with current
practice worldwide, kidney biopsy was not performed and
echocardiography was performed if cardiac examination was
abnormal. Management involved the use of furosemide (1-2 mg/kg
daily, as indicated), enalapril (0.3-0.4 mg/kg/day orally),
intravenous infusions of albumin (1-2 g/kg once every 7-14
days), and supplements of thyroxine (5-10 µg/kg/day) and
vitamins, while ensuring adequate nutrition. Parents were
counselled regarding outcomes including risk of progression to
end stage kidney disease, and families opted for a palliative
care plan due to costs of kidney replacement therapy.
The methodology of NGS, performed at
Institute of Genomic and Integrative Biology, Delhi, is detailed
in Supp. Methods. Whole exome sequencing (WES) was
performed using the Illumina HiSeq2000 or NovaSeq platforms,
sequenced reads were mapped and aligned to the reference genome
(GRCh37; hg19), and called and annotated variants in 89 genes
associated with nephrotic syndrome (Supp. Table SI)
[3,11-14] were prioritized based on rarity (minor allele
frequency, MAF <0.1%), novelty in population databases [15-17],
prediction of deleteriousness by in silico tools, and if
previously reported with disease [18]. Only pathogenic and
likely pathogenic variants, according to criteria of the
American College of Medical Genetics and Genomics (ACMG) 2015
guidelines [18,19] were considered causative, and were validated
by Sanger sequencing. Sanger sequencing on parents’ samples was
used to confirm allele segregation for compound heterozygous
variants. Haplotype studies were performed to determine if the NPHS1 variant
c.2600G>A (p.Gly867Asp) that segregated in 10 of 34
patients occurred on a common genetic background, suggesting
inheritance from a common ancestor (founder mutation) (Supp.
Methods) [20].
Statistical analyses: Data was summarized
as median (interquartile range, IQR) for continuous variables
and percentage with 95% confidence interval (CI) for dichotomous
variables. Assuming 70% prevalence of pathogenic or likely
pathogenic variations in genes encoding key podocyte proteins in
patients with congenital nephrotic syndrome [1,3,12,13], 21
patients were required to be enrolled for a precision of 20%, at
power of 80% and alpha error of 5%.
RESULTS
Samples were collected from 34 unrelated
patients (53% boys) with congenital NS diagnosed at 7 centers
across India. Onset of edema was at median age of 20 (IQR 15-45)
days of life, and was associated with anasarca (91.2%), oliguria
(41.2%), poor feeding (35.3%), seizures (32.3%), hypovolemia
(23.5%), severe infections (20.6%) and/or lethargy (8.8%). Ten
(29.4%) patients were born premature and 13 (38.2%) had low
birth weight (Supp. Table SII). Consanguinity and similar
illness in siblings were reported in 11 (32.4%) cases, each.
Median weight for age standard deviation
score (SDS) was -3.1 (IQR -4.1, -1.9), length for age SDS was
-3.9 (IQR -4.6, -2.2) and head circumference SDS was -3.2 (IQR
-4.5, -2.2). Seven (20.6%) patients had hypertension. Isolated
extrarenal features were observed in 9 patients (Supp. Table
SII), while one patient each had features of Galloway-Mowat
and Pierson syndrome. One patient had albinism and microcephaly
and history of sibling death with similar symptoms.
The median blood level of albumin was 1.2
(IQR 0.9-1.4) g/dL, cholesterol 274 (234-349) mg/dL, creatinine
0.4 (0.3-0.7) mg/dL, and estimated glomerular filtration rate (eGFR)
60 (28.3-96) mL/minute per 1.73 m 2
[21]. Seven (20.6%) patients had eGFR <30 mL/minute per 1.73 m2
at evaluation. Three (8.8%) patients had enlarged kidneys
without hydronephrosis or venous thrombosis. There was no
history of significant teratogenic drug intake during pregnancy
or evidence of intrauterine infection.
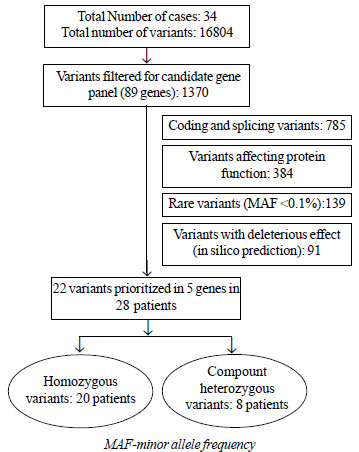 |
|
Fig. 1 Flowchart for variant
filtering after whole exome sequencing.
|
WES with mean coverage of ³30x (Web Table
SIII) returned 16804 variants, of which 1370 variants were
present in one or more of the targeted genes (Fig. 1).
After filtering, 91 variants were shortlisted (Supp. Table
SIV), of which 22 variants were prioritized in 28 patients (Table
I; Supp. Fig. S1). Pathogenic and likely pathogenic
variants were inherited as homozygous and compound heterozygous
variations in 20 and 8 patients, respectively. A monogenic cause
was thus established in 82.4% (95% CI 66.9% to 92.5%) of 34
patients with congenital NS. Most variants were conserved across
species (Web Fig. S2).
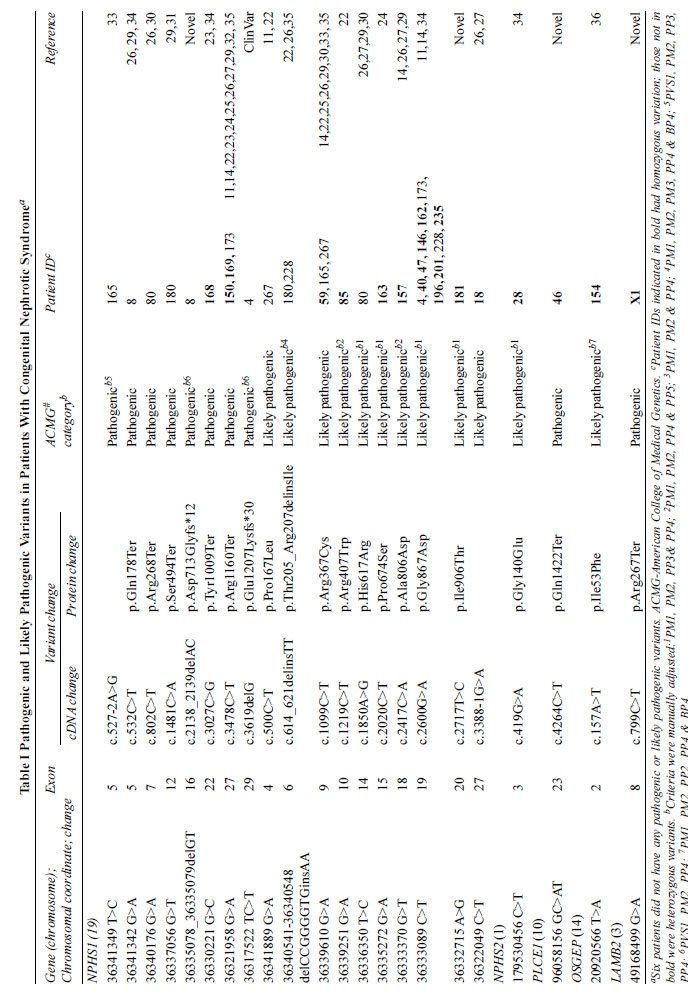 |
Variants in NPHS1 were most common,
including 16 reported [11,12,14,15,22-35] and two novel
variants, segregated in 24 patients as homozygous (n=16)
and compound heterozygous (n=8) variants (Table I).
Reported variations included 7 pathogenic and 9 likely
pathogenic variants. One novel homozygous variant in ID#181 was
classified as likely pathogenic, while another novel NPHS1
variant that segregated as compound heterozygous in ID#8,
was assigned as pathogenic. Fig. 2 indicates the
distribution of defects in NPHS1 across the structure of
nephrin.
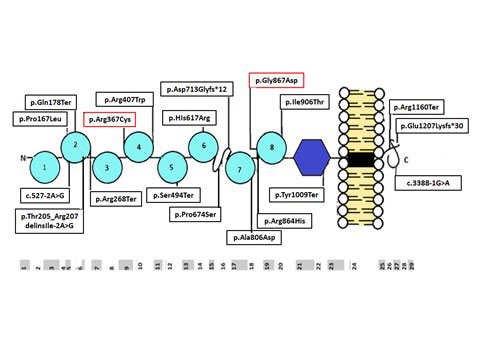 |
|
Fig. 2 Localization of novel
variations and known mutations in the translated nephrin
protein, comprised of eight extracellular immunoglobulin
(Ig) -like domains (semi-circles), a fibronectin type
III-like module (hexagon), a transmembrane domain (black
rectangle) and a C-terminal (C) cytoplasmic domain
(curled line). The bottom panel indicates the exons
coding for the corresponding protein domains. Note that
the 18 variations observed were spread throughout the
protein. The variations with dotted lines are known or
speculated to be founder mutations.
|
One previously reported [11,14] likely
pathogenic NPHS1 variant in exon 19 (c.G2600A;
p.Gly867Asp) was inherited as homozygous in 7 and heterozygous
in 3 patients from different ethnic and regional backgrounds,
without any specific phenotype (Tables I and
SII).
Two other reported variations, p.Arg1160Ter [11,14,27] and
p.Arg367Cys [14,25,27], were common to three patients each (Table
I). In patients with NPHS1 variants, atrial septal
defect was seen in two patients, and developmental delay, facial
dysmorphism, clubbing, café au lait spots, hirsutism and
aqueductal stenosis in one patient each (Supp. Table SII).
One patient each had homozygous likely
pathogenic variants in NPHS2 [34] and OSGEP [36],
associated with an atrial septal defect and Galloway-Mowat
syndrome, respectively. One patient each had novel pathogenic
homozygous variations in PLCE1 and LAMB2 genes;
the latter was associated with phenotype consistent with Pierson
syndrome.
No variants were prioritized in two patients;
four patients had heterozygous variations that were of unknown
significance (Supp. Table SIV). Patients with causative
variations also had additional heterozygous variations (Supp.
Table SIV).
There were no differences in sex ratio, age
at onset of symptoms, levels of serum albumin or estimated GFR
between patients with NPHS1 variations and those with
other or no significant variations (P>0.05 each).
Forty-four of 900 single nucleotide
polymorphisms (SNPs) (Supp. Table SV) in the region (±500
kbp) flanking the c.2600G>A (Gly867Asp) were selected for
haplotype analysis in 33 patients. All 17 alleles carrying the
c.2600G>A variant (homozygous in 7 and heterozygous in 3
patients) shared a core haplotype in the 500 kbp region between
rs2230181 to rs466452 (Supp. Table SVI). Thirteen of 17
alleles also shared a core haplotype extending to 800 kbp
length. The 500 kbp core haplotype was observed in only one of
49 non-mutant chromosomes, suggesting a founder effect.
DISCUSSION
There is significant heterogeneity in
prevalence of inherited defects across studies (Supp. Table
SVII) [6-10,12,22-26,34]. Variants in NPHS1
predominate even in non-Finnish cohorts, and contributions by
NPHS2, WT1 and LAMB2 defects differ widely
across populations. In the present study, the use of NGS enabled
a diagnosis in 82% of 34 patients. These findings are unlike
previous studies from non-Caucasian populations that report
lower rates of inherited defects, perhaps due to focused testing
including a few genes (Supp. Table SVII).
Two founder deletion mutations in NPHS1,
accounting for the majority of cases of Finnish type of
congenital nephrotic syndrome, were not observed in our
patients, similar to reports from non-Finnish populations
[2,3,6-10]. Over 200 NPHS1 mutations are described
worldwide in non-Finnish populations [29,32]. In our report,
homozygous and compound heterozygous mutations in NPHS1
accounted for 70.6% of cases of congenital NS, and 85.7% of
cases with an identified genetic etiology. This proportion is
higher than previous reports from Asia, in which NPHS1
mutations accounted for 22-67% of cases, but similar to
proportions reported in series including non-Finnish populations
(Supp. Table SVII).
As shown in Fig. 2,
variants in NPHS1 were distributed all over the protein.
Three patients shared the variant p.Arg1160Ter, responsible for
premature truncation of protein in the intracellular domain that
interacts with podocin. This variant, a founder mutation in
Maltese patients, is associated with a different allele in Asian
patients [25]. While Koziell, et al. reported a mild phenotype
in affected girl infants [25], we and other authors [24,35]
found a severe phenotype, irrespective of gender,
indistinguishable from other NPHS1 mutations. Three
patients carried a variant (c.1099C>T; p.Arg367Cys), reported
previously as a founder mutation from India [12]. One NPHS1
variant, c.2600G>A (p.Gly867Asp), that translates into a change
in the immunoglobulin-like domain 8, found in 10 unrelated
patients from five states in north India (Supp. Table
SII, Table I and Fig. 1),
has been reported from India, Pakistan and Saudi Arabia
[8,11,14,37], but not from east Asia [6,7,9] or Europe. Using
statistical tools considered more efficient that conventional
haplotyping [20], we show that c.2600G>A is possibly a founder
mutation, as suggested by the lack of genetic variation in the
500-800 kbp length flanking regions [38]. The differences in
frequency of the shared haplotype in various ethnicities in the
1000 genome database suggests a European origin for the mutation
(Supp. Table SVIII) [15]. Our hypothesis requires
confirmation by examining for the same shared haplotype in
previously reported patients with the p.Gly867Asp mutation.
Mutations in NPHS2 and WT1
account for 0-51% and 0-40% cases, respectively, across
populations, though NPHS2 variants are uncommon in Asia (Supp.
Table SVII). In this cross-sectional study, only one
patient had homozygous mutations in NPHS2, and none had
variants in WT1. Given the small study size, these
findings have limited generalisability.
Confirming previous findings, we failed to
find specific phenotypic associations in patients with NPHS1,
NPHS2 and PLCE1 mutations [4,26,39]. The lone
patient with homozygous LAMB2 variant had findings of
Pierson syndrome while another had Galloway-Mowat syndrome
secondary to OSGEP mutation [36]. The latter patient had
the same mutation and phenotype as an infant of Pakistani
ethnicity described previously [36].
The present series underscores the utility of
providing a genetic etiology in patients with congenital NS,
thereby facilitating prenatal counseling and testing in
subsequent pregnancies. One NPHS1 mutation is
hypothesized to have a founder effect in Indian population.
Information on long term outcomes, including
post-transplantation, is lacking since most children were lost
to follow up after families chose a palliative care plan.
Despite being a multicenter study, the findings of the
relatively small sample size might not be generalizable.
Note: Supplementary material related to
this study is available with the online version at
www.indianpediatrics.net
Acknowledgments: We thank the following
colleagues participating in the NephQuest network who
contributed samples and details of their patients: SP Veeturi,
Rainbow Children Hospital, Hyderabad; KL Tiewsoh, Postgraduate
Institute of Medical Education and Research, Chandigarh; A
Mittal, All India Institute of Medical Sciences, Jodhpur; S
Krishnamurthy, Jawaharlal Institute of Postgraduate Medical
Education and Research, Puducherry; M Mantan, Maulana Azad
Medical College, New Delhi; M Kumar and K Mishra, Chacha Nehru
Bal Chikitsalaya, Delhi.
Ethics approval: Ethics committees at
CSIR Institute of Genomics and Integrative Biology, Delhi and
All India Institute of Medical Sciences, New Delhi; Sanction no.
IECPG-616/21.12.2016. RT-33/22.03.2017 and
6/GAP127/CSIR-IGIB/2017
Contributors: All authors
contributed to the study conception and design. AJ, AS, AS, MF,
AB: material preparation, data collection and analysis were
performed; AJ, AS: The first draft of the manuscript was written
jointly. All authors commented on the manuscript, and approved
the final manuscript.
Funding: Department of Biotechnology,
Government of India (BT/11030/MED/30/1644/2016).
Competing interest: None stated.
| |
|
WHAT IS ALREADY KNOWN?
• Genetic defects account for 60-80%
of cases with congenital nephrotic syndrome
• Mutations in NPHS1 are most
common in Caucasians; WT1 and LAMB2
variants are probably more common in Asian patients
WHAT THIS STUDY ADDS?
• Genetic defects are present in more
than 80% patients with congenital nephrotic syndrome in
India
• Mutations in NPHS1 account
for more than 80% of patients with an inherited basis
• Common variants in NPHS1 are those that are
known (c.1099C>T; p.Arg367Cys) or speculated (c.2600G>A;
p.Gly867Asp) to be founder mutations.
|
REFERENCES
1. Jalanko H. Congenital nephrotic syndrome.
Pediatr Nephrol 2009; 24: 2121-28
2. Norio R. Heredity in the congenital
nephrotic syndrome. A genetic study of 57 Finnish families with
a review of reported cases. Ann Paediatr Fenn 1996;12:S27:1-94
3. Ha TS. Genetics of hereditary nephrotic
syndrome: a clinical review. Korean J Pediatr 2017;60:55-63
4. Hinkes BG, Mucha B, Vlangos CN, et al;
Arbeitsgemeinschaftfür Paediatrische Nephrologie Study Group.
Nephrotic syndrome in the first year of life: Two thirds of
cases are caused by mutations in 4 genes (NPHS1, NPHS2, WT1, and
LAMB2). Pediatrics 2007;119: e907-19
5. Wang JJ, Mao JH. The etiology of
congenital nephrotic syndrome: Current status and challenges.
World J Pediatr 2016;12:149-58
6. Li GM, Cao Q, Shen Q, et al. Gene mutation
analysis in 12 Chinese children with congenital nephrotic
syndrome. BMC Nephrol 2018;19:382.
7. Sako M, Nakanishi K, Obana M, et al.
Analysis of NPHS1, NPHS2, ACTN4, and WT1 in Japanese patients
with congenital nephrotic syndrome. Kidney Int 2005;67: 1248-55
8. Sharief SN, Hefni NA, Alzahrani WA, et al.
Genetics of congenital and infantile nephrotic syndrome. World J
Pediatr 2019;15:198-203
9. Nishi K, Inoguchi T, Kamei K, et al.
Detailed clinical manifestations at onset and prognosis of
neonatal-onset Denys-Drash syndrome and congenital nephrotic
syndrome of the Finnish type. Clin Exp Nephrol 2019;23:1058-65
10. Sinha R, Vasudevan A, Agarwal I, et al.
Congenital nephrotic syndrome in India in the current era: A
multicenter case series. Nephron 2019;144:1-9
11. Bierzynska A, McCarthy HJ, Soderquest K,
et al. Genomic and clinical profiling of a national nephrotic
syndrome cohort advocates a precision medicine approach to
disease management. Kidney Int 2017;91:937-47
12. Sadowski CE, Lovric S, Ashraf S, et al;
SRNS Study Group, Hildebrandt F. A single-gene cause in 29.5% of
cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol
2015;26:1279-89
13. Trautmann A, Lipska-Ziêtkiewicz BS,
Schaefer F. Exploring the clinical and genetic spectrum of
steroid resistant nephrotic syndrome: The PodoNet registry.
Front Pediatr 2018;6:200
14. Lovric S, Fang H, Vega-Warner V, et al;
Nephrotic Syndrome Study Group. Rapid detection of monogenic
causes of childhood-onset steroid-resistant nephrotic syndrome.
Clin J Am Soc Nephrol 2014;9:1109-16
15. 1000 Genomes Project Consortium; Auton A,
Brooks LD, Durbin RM, et al. A global reference for human
genetic variation. Nature 2015; 526: 68-74
16. Karczewski KJ, Weisburd B, Thomas B, et
al; The Exome Aggregation Consortium, Daly MJ, MacArthur DG. The
ExAC browser: Displaying reference data information from over
60000 exomes. Nucleic Acids Res 2017;45 (D1): D840-5
17. Karczewski KJ, Francioli L, Tiao G, et
al. The mutational constraint spectrum quantified from variation
in 141,456 humans. Nature 2022;581:434-43.
18. Hamosh A, Scott AF, Amberger JS, Bocchini
CA, McKusick VA. Online Mendelian Inheritance in Man (OMIM), a
knowledgebase of human genes and genetic disorders. Nucleic
Acids Res 2005;33:D514-7
19. Richards S, Aziz N, Bale S, et al; ACMG
Laboratory Quality Assurance Committee. Standards and Guidelines
for the Interpretation of Sequence Variants: A Joint Consensus
Recommendation of the American College of Medical Genetics and
Genomics and the Association for Molecular Pathology. Genet Med
2015;17:405-24
20. Stephens M, Smith NJ, Donnelly P. A new
statistical method for haplotype reconstruction from population
data. Am J Hum Genet 2001;68:978-89
21. Schwartz GJ, Muñoz A, Schneider MF, et
al. New equations to estimate GFR in children with CKD. J Am Soc
Nephrol 2009;20:629-37
22. Schoeb DS, Chernin G, Heeringa SF, et al;
Gesselschaftfür Paediatrische Nephrologie Study Group. Nineteen
novel NPHS1 mutations in a worldwide cohort of patients with
congenital nephrotic syndrome (CNS). Nephrol Dial Transplant
2010;25:2970-6
23. Lee JH, Han KH, Lee H, et al.
Genetic basis of congenital and infantile nephrotic syndromes.
Am J Kidney Dis 2011;58:1042-3
24. Warejko JK, Tan W, Daga A, et al. Whole
exome sequencing of patients with steroid-resistant nephrotic
syndrome. Clin J Am Soc Nephrol 2018;13:53-62
25. Koziell A, Grech V, Hussain S, et al.
Genotype/phenotype correlations of NPHS1 and NPHS2 mutations in
nephrotic syndrome advocate a functional inter-relationship in
glomerular filtration. Hum Mol Genet 2002;11:379-88
26. Machuca E, Benoit G, Nevo F, et al.
Genotype-phenotype correlations in non-Finnish congenital
nephrotic syndrome. J Am Soc Nephrol 2010;21:1209-17
27. Lenkkeri U, Männikkö M, McCready P, et
al. Structure of the gene for congenital nephrotic syndrome of
the Finnish type (NPHS1) and characterization of mutations. Am J
Hum Genet 1999;64:51–61
28. Kestilä M, Lenkkeri U, Männikkö M, et al.
Positionally cloned gene for a novel glomerular protein-nephrin-is
mutated in congenital nephrotic syndrome. Mol Cell 1998;1:575-82
29. Beltcheva O, Martin P, Lenkkeri U,
Tryggvason K. Mutation spectrum in the nephrin gene (NPHS1) in
congenital nephrotic syndrome. Hum Mutat 2001;17: 368-73
30. Ulinski T, Aoun B, Toubiana J, Vitkevic
R, Bensman A, Donadieu J. Neutropenia in congenital nephrotic
syndrome of the Finnish type: Role of urinary ceruloplasmin
loss. Blood 2009;113:4820-1
31. Bolk S, Puffenberger EG, Hudson J, Morton
DH, Chakravarti A. Elevated frequency and allelic heterogeneity
of congenital nephrotic syndrome, Finnish type, in the old order
Mennonites. Am J Hum Genet 1999; 65: 1785-90
32. Heeringa SF, Vlangos CN, Chernin G, et
al; Members of the APN Study Group. Thirteen novel NPHS1
mutations in a large cohort of children with congenital
nephrotic syndrome. Nephrol Dial Transplant 2008;23:3527-33
33. Ashton E, Bockenhauer D, Boustred C,
Jenkins L, Lench N. ASHG 2013 Poster: Development of a rapid
genetic testing service for nephrotic syndrome using next
generation sequencing. Conference: American Society of Human
Genetics, At Boston, USA 2013; available at
https://www.researchgate.net/publication/258860074_ASHG_
2013_Poster_Development_of_a_rapid_genetic_ testing_
service_for_nephrotic_syndrom e_using_next_generation_
sequencing; last accessed 21 February 2020
34. Sen ES, Dean P, Yarram-Smith L, et al.
Clinical genetic testing using a custom-designed
steroid-resistant nephrotic syndrome gene panel: Analysis and
recommendations. J Med Genet 2017;54:795-804.
35. Ovunc B, Ashraf S, Vega-Warner V, et al;
Gesellschaft für Pädiatrische Nephrologie (GPN) Study Group.
Mutation analysis of NPHS1 in a worldwide cohort of congenital
nephrotic syndrome patients. Nephron Clin Pract 2012;120:c139-46
36. Domingo-Gallego A, Furlano M, Pybus M, et
al. Novel homozygous OSGEP gene pathogenic variants in two
unrelated patients with Galloway-Mowat syndrome: Case report and
review of the literature. BMC Nephrol 2019;20:126
37. Abid A, Khaliq S, Shahid S, et al. A
spectrum of novel NPHS1 and NPHS2 gene mutations in pediatric
nephrotic syndrome patients from Pakistan. Gene 2012;502:133-7
38. Zlotogora J. High frequencies of human
genetic diseases: founder effect with genetic drift or
selection? Am J Med Genet 1994;49:10-3
39. Schultheiss M, Ruf RG, Mucha BE, et al.
No evidence for genotype/phenotype correlation in NPHS1 and
NPHS2 mutations. Pediatr Nephrol 2004;19:1340-8.
|
|
|
 |
|
