Nephrocalcinosis (NC) is defined as calcium deposition in
the renal parenchyma as detected by renal ultrasonogram.
Pediatric NC is a rare entity and might occur secondary to
inherited renal tubular disorders, vitamin D excess, etc [1-4].
A comprehensive metabolic evaluation of NC would help in
specific therapies, prevent progression to end-stage renal
disease and enable optimal prenatal counseling.
Most published information on NC is from developed nations [1,5]
and there is paucity
of information regarding pediatric NC from India [6]. Since NC
often has an underlying genetic or metabolic etiology, it can be
speculated that its etiological profile is likely to vary with
ethnicity. We
studied the etiological profile of NC among children from
Southern India.
METHODS
This
cross-sectional study was conducted at the pediatric nephrology
clinic of a referral hospital in Southern India from July, 2017
through July, 2019, after obtaining approval from the
institutional ethics committee. Prior written informed consent
was obtained from the parents. The primary objective of the
study was to evaluate the underlying etiology of NC, while the
secondary objectives were to record the patterns of clinical
presentation, complications and outcomes (estimated glomerular
filtration rate (eGFR) on follow-up) in these children.
All patients
≤18 years with NC who were referred for
diagnostic evaluation were included. NC was defined as calcium
deposition in the renal parenchyma as detected and graded by
ultrasound. Medullary NC was graded as: grade 1, mild increase
in echogenicity of medullary pyramids; grade 2, mild diffuse
increase in echogenicity of medullary pyramids without acoustic
shadowing; and grade 3, greater homogenous increase in
echogenicity of medullary pyramids with acoustic shadowing [1].
Cortical nephrocalcinosis was diagnosed by the presence of
calcifications in the renal cortex.
This was an observational study, supplemented by analysis of
hospital records. For the prospective component of the study,
consecutively presenting children who were referred for
evaluation of NC were evaluated. For the retrospective component
of the study, data were collected from the records of children
18 years or younger with NC, who had presented to the pediatric
nephrology clinic over the last 10 years and were under
follow-up at the pediatric nephrology clinic.
We have been using the following protocol for investigating NC
for the last 10 years: (i) First-line investigations-
Blood pH, blood urea, creatinine, sodium, potassium, magnesium,
chloride, serum bicarbonate, calcium, phosphorous, alkaline
phosphatase, uric acid; urinalysis for urine pH, crystals, urine
culture (if clinically indicated), spot calcium: creatinine
ratio and 24-hour urine excretion of calcium, oxalate, uric acid
and creatinine, were recorded. Estimated glomerular filtration
rate (eGFR) was determined using modified Schwartz formula [7].
(ii) Second-line investigations- Urine sodium
nitroprusside test was restricted to patients where a cause of
NC was not found on first-line investigations. Blood parathyroid
hormone (PTH) and 25 hydroxy-cholecalciferol was evaluated in
patients with hypercalcemia (serum calcium >11mg/dL on >2
occasions). Urine
b2 microglobulin levels were performed in males
with suspected Dent disease.
Following definitions were used for defining the etiology of
nephrocalcinosis [6, 8-10]: Distal renal tubular acidosis (RTA)
was diagnosed in patients with suggestive clinical features
(failure to thrive, polyuria, rickets, hypokalemic paralysis,
etc) and hyperchloremic metabolic acidosis (serum
bicarbonate <18 mEq/L), normal anion gap (8-12 mEq/L), normal
fractional excretion of bicarbonate (<5%), urine pH >5.5 and
hypercalciuria (elevated urinary calcium >4 mg/kg per day in a
24 hour urine sample). Idiopathic hypercalciuria was defined as
hypercalciuria with absence of other tubular defects and
normocalcemia (9-11 mg/dL). Bartter syndrome was diagnosed in
children with suggestive clinical features (failure to thrive,
polyuria, etc), metabolic alkalosis (serum bicarbonate >25
mEq/L), hypokalemia (potassium <3.5 mEq/L), normal blood
pressure, increased urinary potassium (>20 mEq/L) and chloride
(>30 mEq/L), with high plasma renin activity. Primary
hyperoxaluria was defined as elevated urinary oxalate excretion
(>40 mg/1.73 m2 per
day on a 24-hour urinary specimen) and no history of
malabsorption, steatorrhea or intestinal surgical resection.
Hyperpara-thyroidism was diagnosed in those with high serum
calcium (>11 mg/dL) and PTH (>50 pg/mL) with or without
hypercalciuria. Hyperuricosuria was diagnosed if uric acid
excretion was >815 mg/1.73m2 on
a 24-hour urine specimen. Dent disease was diagnosed as per
standard definitions [10]. Familial hypomagnesemia
with hypercalciuria and nephrocalcinosis (FHHNC)
was diagnosed in those with low serum magnesium
(<1.5 mg/dL), urinary magnesium wasting (fractional
excretion
≥5%),
hypercalciuria, NC with/without family history of hypomagnesemia
with hypercalciuria. Idiopathic hypercalcemia of infancy was
diagnosed when hypercalcemia (>11 mg/dL) was noted in the
absence of vitamin D toxicity, hyper-parathyroidism, absence of
calcium supplement intake or subcutaneous fat necrosis.
Determination of the cause of NC was followed by specific
therapy. Patients were advised to consume plenty of fluids and
to restrict intake of added salts. Long term outcome was
assessed in terms of clinical improvement, weight Z
score, height Z score and renal functions.
Statistical analyses: The data were
analyzed by SPSS 23.0. Normality of data was analyzed by
Kolmogorov-Smirnov test.
Paired t test was used to compare means of two dependent
sample groups. Median and IQR of two dependent sample groups
were compared using Wilcoxon-signed rank sum test.
RESULTS
Of the 54 children with NC (29 males), 18 were recruited
prospectively. Fifty-two children had medullary NC. One child
with primary hyperoxaluria had both cortical and medullary NC,
while 1 child with autosomal recessive polycystic kidney disease
(ARPKD) had cortical NC. Fig. I shows the
etiological profile of NC in our study. Distal RTA, primary
hyperoxaluria, Bartter syndrome and Dent disease were the most
common causes of NC.
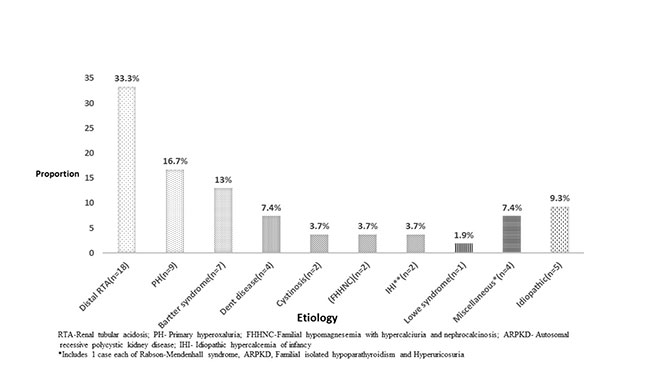 |
| Fig. 1 Etiology of
nephrocalcinosis in the enrolled children (N=54). |
Dent disease was diagnosed in 4 cases, of which 2 cases had type
1 phenotype (with no metabolic acidosis), while the other 2
cases had a phenotype consistent with type 2 Dent disease (with
metabolic acidosis). Out of two children with FHHNC, one had
positive family history of hypomagnesemia and urolithiasis in a
maternal uncle. Cystinosis was diagnosed in two cases of NC, who
had Fanconi syndrome and cystine crystals in cornea.
They were treated with potassium citrate, phosphorus
supplements and oral cysteamine (in one case). Another child
with medullary NC had global developmental delay, bilateral
cataracts, hypotonia and Fanconi syndrome; and was diagnosed as
Lowe syndrome. Two infants were diagnosed as idiopathic
hypercalcemia of infancy (serum calcium 12.5 mg/dL and 12 mg/dL,
respectively) and were treated with bisphosphonates, on which
the serum calcium levels normalized.
Table I Baseline Clinical and Biochemical Characteristics of Children with Nephrocalcinosis (N=54)
|
Parameters |
Value |
|
Age at symptom onset, mo |
24 (6, 48) |
|
#Age at diagnosis, mo |
36 (11.5, 84) |
|
#Symptom-diagnosis interval, mo |
24 (7.6, 59) |
|
‡Clinical features | |
|
Failure to thrive (WFA<-2 Z score) |
29 (53.7) |
|
Polyuria |
24 (44.4) |
|
Polydipsia |
17 (31.5) |
|
Rickets |
19 (35.2) |
|
Hypokalemic paralysis |
6 (11.1) |
|
Short stature |
5 (9.3) |
|
Carpopedal spasm |
5 (9.3) |
|
Pathological fractures |
4 (7.4) |
|
Acute kidney injury |
3 (5.6) |
|
Hematuria |
4 (7.4) |
|
Biochemical features | |
|
*eGFR at presentation, mL/min/1.73m² |
59 (25.5) |
|
Metabolic acidosis |
26 (48.1) |
|
#Serum creatinine at presentation, mg/dL |
0.59 (0.49, 0.70) |
|
eGFR <60 mL/min/1.73 m2 at diagnosis |
24 (44.4) |
|
eGFR <60 mL/min/1.73 m2 at last follow up |
15 (27.8) |
|
^Associated urolithiasis |
3 (5.55 |
|
Values in no. (%) except *mean (SD) or #median (IQR); ‡Recurrent vomiting, salt craving, and antenatal detection in 2 each; eGFR-Estimated Glomerular filtration rate, WFA- Weight for age; ^One child each with primary hyperoxaluria, familial hypomagnesemia and hypercalciuria with nephrocalcinosis (FHHNC), and Rabson-Mendenhall syndrome. |
)
Table
I
provides the baseline clinical and biochemical features of the
enrolled children.
Grade 1, 2 and 3 nephrocalcinosis were noted in 45 (83.3%), 8
(14.8%) and 1 (1.9%) of cases, respectively. All the NC cases
(except the 5 idiopathic cases) had hypercalciuria. There was
history of consanguinity in 27 (50%) of cases, while there was a
family history of nephrocalcinosis in 14 (25.9%) of cases. The
mean (SD) 24-hour urinary oxalate in 9 children with primary
hyperoxaluria was 85 (31.8) mg/1.73 m2/day.
Four children presented with persistently low eGFR for more than
3 months. During the median (IQR) duration of follow up of 24
(8, 56) months in children with NC, there was improvement in the
weight Z scores and eGFR (Table II).
Table II Growth and Biochemical Features at Presentation and at Follow-up in Children with Nephrocalcinosis (N=54)
|
Parameter |
At presentation |
At last follow up† |
|
*Age. mo |
36 (11.5, 84) |
78 (38, 144) |
|
*Weight (Z score) |
-3.4 (-2.0, -4.98) |
-2.95(-3.75,-1.68) |
|
#Height (Z score) |
-3.20 (2.04) |
-2.96 (2.02) |
|
#eGFR*, mL/min/1.73m2 |
59.04 (25.5) |
77 (31.48) |
|
†Median (follow up) of 24 (8,56) mo; eGFR-estimated Glomerular filtration rate using modified Schwartz formula; values in *median (IQR) or #mean (SD);P<0.01 for all comparisions except age. |
Genetic studies were performed in 8 children. Out of these, in
five children with
primary hyper-oxaluria, AGXT mutation was detected in four
cases; and GRHPR mutation in one. One child with distal RTA had
ATP6V0A4 mutation, and two children with Bartter syndrome had
ROMK and CLCN-KB mutations, respectively.
DISCUSSION
This study is one
of the largest single-centre studies on the etiological profile
of NC. The study showed that the most common etiologies of NC
were distal RTA, primary hyperoxaluria, Bartter syndrome and
Dent disease, together accounting for more than two-thirds of
cases. Common clinical presentations included failure to thrive,
polyuria and bony deformities. At a median (IQR) follow up of 24
(18, 56) months, the estimated glomerular filtration rate (GFR)
had significantly increased, possibly due to resolution of AKI
(resulting from a polyuric state).
There have been few studies
evaluating the clinico-etiological profile of pediatric NC
[1,4-6, 11]. Mantan, et al. [6] retrospectively evaluated
the etiology of NC in 40 children from northern India, which
included d-RTA (50%), idiopathic hypercalciuria (7.5%) and
primary hyperoxaluria (7.5%). At a median (range) follow up of
35 (14,240) months, the eGFR had declined from 82.0 (42,114) to
70.8 (21.3, 126.5) mL/min/1.73 m2.
Ronnefarth, et al., [1] retrospectively evaluated 152
children with NC from Germany, which included idiopathic
hypercalciuria (34%), hereditary tubular disorders (32%) and
vitamin D toxicity (8%). The eGFR had increased from 96 to 103
mL/min/1.73m2.
Dogan, et al., [4] in 36 Turkish children with NC,
reported distal RTA (30.5%), Bartter syndrome (13.8%), Vitamin D
toxicity (8.3%), idiopathic hypercalciuria (5.5%) and primary
hyperoxaluria (5.5%).
Among 41 children from Italy, hereditary tubulopathies
was the single largest etiology (41.4%), of which distal RTA was
seen in 17% [5]. During a mean follow up of 4.4 years, eGFR
remained stable in 89% [5].
There appear to be some differences
in our results when compared to those of the aforementioned
studies [1,4,6]. The etiological profile of our enrolled cases
is notable for the absence of idiopathic hypercalciuria, which
in previous studies ranged from 7.5%-34% [1,4,6]. The
hypercalciuria in our enrolled cases was secondary in nature.
Furthermore, a cause for NC was not identifiable in 9.3% of
enrolled cases in our study. This is comparable to the results
of the Arbeitsgemeinschaft für Pädiatrische Nephrologie (APN)
survey (6%) [1]. Consanguinity was noted in 50% of our cases;
and this, along with ethnic variations could have accounted for
high percentage of inherited tubulopathies. Hypercalciuria was
commonly noted in our study, highlighting its importance as a
major pathogenic factor [12,13].
Primary hyperoxaluria was another important cause,
similar to those reported in developed countries [14,15]. We did
not encounter any cases of vitamin D excess among the enrolled
cases.
Owing to resource constraints,
genetic studies could not be performed in all cases.
Moreover, we could not perform urine citrate estimation
due to logistic reasons. Finally, the etiological profile of
patients enrolled in this study might be affected by a referral
bias.
To summarize, distal RTA, primary
hyperoxaluria, Bartter syndrome and Dent disease were the most
common etiologies of NC in our study. Failure to thrive,
polyuria, polydipsia and bony deformities were the common
presenting features in our patients. With a systematic approach,
etiologies of NC could be identified in most of the cases.
Contributors:
KR, SK, PS: management of the patients; KR: collected the
data, reviewed the literature and drafted the first version of
the manuscript; SK: conceptualized the study, collected the
data, reviewed the literature, revised the manuscript and
critically reviewed the manuscript. All authors contributed to
drafting of the manuscript and approved the final version of the
manuscript; SK: shall act as guarantor of the paper.
Funding:
Supported, in part, by institutional and departmental funds.
Competing interests:
None stated.
WHAT THIS STUDY ADDS?
• Distal renal tubular acidosis, primary
hyperoxaluria, Bartter syndrome and Dent disease were
the commonest etiologies of nephrocalcinosis at a
referral hospital in Southern India. |
REFERENCES
1. Ronnefarth G, Misselwitz
J. Nephrocalcinosis in children: A retrospective survey. Pediatr
Nephrol. 2000;14:1016-21.
2. Lin MT, Tsau YK, Tsai
WY, Tsai WS, Lu FL, Hsiao PH, et al.
Nephrocalcinosis in childhood. Acta Paediatr Taiwanica. 1999;
40:27-30.
3. Manz F, Jaschke W, van
Kaick G, Waldherr R, Willich E. Nephrocalcinosis in radiographs,
computed tomography, sonography and histology. Pediatr Radiol.
1980;9:19-26.
4. Doðan CS, Uslu-Gökçeoðlu
A, Comak E, Alimoðlu E, Koyun M, Akman S. Renal function and
linear growth of children with nephrocalcinosis: A retrospective
single-center study. Turk J Pediatr. 2013; 55:58-62.
5. Ammenti A, Pelizzoni A,
Cecconi M, Molinari PP, Montini G. Nephrocalcinosis in children:
a retrospective multi-centre study. Acta Paediatr.
2009;98:1628-31.
6. Mantan M, Bagga A, Virdi
VS, Menon S, Hari P. Etiology of nephrocalcinosis in northern
Indian children. Pediatr Nephrol. 2007;22:829-33.
7. Schwartz GJ, Work DF.
Measurement and estimation of GFR in children and adolescents.
Clin J Am Soc Nephrol. 2009;4:1832-43.
8. Habbig S, Beck BB, Hoppe
B. Nephrocalcinosis and urolithiasis in children. Kidney Int.
2011;80:1278-91.
9. Cameron MA, Sakhaee K,
Moe OW. Nephrolithiasis in children. Pediatr Nephrol. 2005;
20:1587-92.
10. Bhardwaj S, Thergaonkar
R, Sinha A, Hari P, Hi C, Bagga A.
Phenotype of Dent disease in a cohort of Indian children.
Indian Pediatr. 2016;53:977-82.
11. Al-Bderat JT, Mardinie
RI, Salaita GM, Al-Bderat AT, Farrah MK. Nephrocalcinosis among
children at King Hussein medical center: Causes and outcome.
Saudi J Kidney Dis Transplant. 2017;28:1064-8.
12. Eggert P, Müller D,
Schröter T.
Nephrocalcinosis in three siblings with idiopathic
hypercalciuria. Pediatr Nephrol.1998;12:144-6.
13. Moxey-Mims MM,
Stapleton FB.
Hypercalciuria and nephrocalcinosis in children. Curr Opin
Pediatr. 1993;5:186-90.
14. Lieske JC, Monico CG,
Holmes WS, Bergstralh EJ, Slezak JM, Rohlinger AL, et al.
International registry for primary hyperoxaluria. Am J Nephrol.
2005;25:290-6.
15. van Woerden CS, Groothoff JW, Wanders RJA,
Davin J-C, Wijburg FA. Primary hyperoxaluria type 1 in The
Netherlands: prevalence and outcome. Nephrol Dial Transplant.
2003;18:273-9.


