|
|
|
Indian Pediatr 2013;50: 510-512 |
 |
Johanson-Blizzard Syndrome
|
|
Koumudi Godbole, *Sukalo Maja,
†Hiremath
Leena and *Zenker Martin
From the Department of Genetic Medicine, Deenanath
Mangeshkar Hospital and Research Center, Erandawane, Pune, India; *Institute
of Human Genetics, University Hospital Magdeburg, Germany and
†Consultant, Department of Pediatrics, Jehangir Hospital, Pune, India.
Correspondence to: Dr K. Godbole, Consultant Clinical
Geneticist, Department of Genetic Medicine, Deenanath Mangeshkar
Hospital and Research Center, Erandawane, Pune 411 004, India.
Email: [email protected]
Received: November 27, 2012;
Initial review: December 10, 2012;
Accepted: December 20, 2012.
|
|
We present clinical features and genetic diagnosis in an Indian infant
diagnosed with Johanson- Blizzard syndrome. This is a rare, autosomal
recessive genetic condition with multi-system involvement and a
characteristic facies. Molecular genetic testing is important to confirm
the clinical diagnosis and offer prenatal diagnosis in future
pregnancies.
Key words: Johanson-Blizzard syndrome, India.
|
Johanson-Blizzard syndrome (MIM2G3800)
is a rare, autosomal recessive genetic condition with a
characteristic ‘diagnostic facies’. We present an Indian
infant with this condition.
Case Report
A 12-day-old female newborn was referred
for Genetics consultation for her unusal facies and
congenital heart defect. She was the first born to 3rd
degree consanguineous parents without any family history of
major medical or genetic disorders, except for
well-controlled maternal hypothyroidism. She was born
normally at term with a birthweight of 2.45 kg and suffered
from a secondary apnea requiring resuscitation followed by
feeding difficulties and poor weight gain.
On examination, on day 12, she weighed
2.3 kg, had head circumference of 32 cm and length of 50 cm.
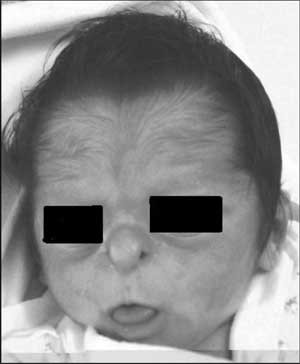
She had a striking facies (Fig. 1) with a
small beak-like nose with hypoplastic alae nasi, long narrow
upper lip, open mouth with protruding tongue, prominent eyes
with palpebral fissures slanting upwards and epicanthic
folds. She had a frontal upsweep of hair with hypertrichosis,
especially on forehead. Additionally, she had clinodactyly
of 5 th fingers
bilaterally. There was a systolic murmur on auscultation and
she was noted to be hypotonic and lethargic. The rest of the
systemic examination was normal. Her echocardiogram revealed
an atrial septal defect with persistent ductus arteriosus
while her abdominal ultrasound was normal. She was reported
to have normal TSH and a normal 46, XX karyotype.
Additionally otoacoustic emission screening had reported
sensorineural hearing loss which was confirmed by BERA test
later. A clinical diagnosis of Johanson-Blizzard syndrome
was considered and molecular genetic testing including
sequencing of UBR1 gene was
done homozygous mutation in the UBR1 gene was
detected.
 |
|
Fig. 1 Patient at age of
12 days: Characteristic facies with beaked nose,
hypoplastic alae nasi, long philtrum with thin upper
lip and frontal hirsutism with upsweep of hair.
|
On review at 7 months of age, pancreatic
insufficiency with excess fat globules in stool sample and
hypothyroidism had been diagnosed. She was receiving
thyroxin supplement and pancreatic supplements for
malabsorption secondary to pancreatic insufficiency with
some improvement in weight (6.1 kg). She was prescribed
bilateral hearing aids and regular physiotherapy with early
intervention program for her developmental delay. Parents
reported repeated hospital admissions for recurrent
respiratory infections.
Molecular analysis
DNA from peripheral blood was extracted
using standard method. All 47 exons including the flanking
intron regions of UBR1 gene were amplified by PCR.
PCR amplicons were purified and subjected to direct
sequencing using an automated sequencer. Sequences were
compared to the reference sequences deposited in the public
database (NM_174916).
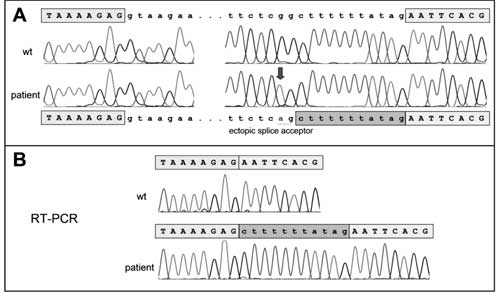
Homozygosity for a nucleotide
substitution in intron 4 of the UBR1 gene was found.
The identified change c.529-13G>A has not been published, so
far, but it was previously discovered in our laboratory in
patient of Hispanic origin with the syndrome. The G to A
substitution was demonstrated to introduce an ectopic splice
site 11 base pairs upstream of the authentic splice acceptor
of exon 5, thus leading to a shift of the reading frame
(p.N177Lfs*10) (Fig. 2 ). This change can
therefore be assumed to represent a disease-causing
mutation.
 |
|
Fig. 2 Electropherograms
derived from gDNA and cDNA sequencing.
|
Discussion
JBS in its typical expression can be
diagnosed at birth with its characteristic facies.
Ultrasound-based prenatal diagnosis has been reported
previously [4,5] suspected by a beak-like nose and a dilated
sigmoid colon suggestive of imperforate anus at 21 weeks of
gestation. No such prenatal ultrasound features were
detected in our patient and ultrasound may not be the best
modality to make a specific diagnosis of JBS especially in
absence of a family history.
Patients with JBS need long-term care
including management of pancreatic insufficiency and
hypothyroidism, treatment of frequent respiratory
infections, management of hearing loss, physiotherapy and
educational rehabilitation depending on the intelligence
level. Pancreatic insufficiency and severe hypoproteinemia
may lead to death in infancy or early childhood, but for
patients managed appropriately, survival into adulthood is
not rare.
UBR1 gene located on chromosome
15q15.2 is currently the only gene associated with JBS. It
encodes E3 ubiquitin ligase of the N-end rule pathway which
is an ubiquitin (Ub) dependent proteolytic pathway. UBR1
is essential in the development and maintenance of acinar
cells and in-utero destruction of acinar tissue
followed by fatty replacement as well as eventual
progression to endocrine deficiency leading to diabetes in
older children has been previously reported [7-9].
Most patients have biallelic mutations
predicting complete loss of function while cases with
missense mutations or small in-frame deletions proposed to
be hypomorphic mutations have been described with somewhat
milder phenotypes and normal intelligence [3]. Our patient
has an intronic mutation close to but not directly affecting
the splice acceptor site of exon 5. We could demonstrate
that this alteration creates an ectopic splice site
resulting in an inclusion of 11 nucleotides from intron 4
into the coding sequence; those 11 additional base pairs
cause a frameshift that lead to a premature stop codon
(p.N177Lfs*10). No evidence of a normally spliced
transcript could be found by RNA analysis. We therefore
presume that this mutation leads to a complete or
near-complete loss of function which is in line with the
classical JBS phenotype seen in this girl.
It is important for pediatricians to
consider molecular testing of UBR1 gene not only for
the confirmation of diagnosis in the affected child but also
for confirming carrier status in both parents and to offer
appropriate counseling to the family.
Contributors: KG and LH were involved
in the diagnosis and management writing the manuscript. MS
and MZ performed laboratory analysis, critically reviewed
the manuscript and also helped in writing it. The final
manuscript was approved by all authors.
Funding: None; Competing interests:
None stated.
References
1. Johanson A, Blizzard R. A syndrome of
congenital aplasia of the alae nasi, deafness,
hypothyroidism, dwarfism, absent permanent teeth and
malabsorption. J Pediatr. 1971;79: 982-7.
2. Moeschler JB, Lubinsky MS. Johanson-Blizzard
syndrome with normal intelligence. Am J Med Genet.
1985;22:69-73.
3. Hwang C-S, Sukalo M, Batygin O, Addor
MC, Brunner H, Aytes AP, et al. Ubiquitin ligases of
the N-end rule pathway: assessment of mutations in UBR1 that
cause the Johanson-Blizzard syndrome. PLoS ONE.
2011;6:e24925.
4. Vanlieferinghen P, Gallot D,
Francannet Ch, Meyer F, Dechelotte P. Prenatal
ultrasonographic diagnosis of a recurrent case of Johanson-Blizzard
syndrome. Genet Couns. 2003;14:105-7.
5. Auslander R, Nevo O, Diukman R, Morrad
E, Bardicef M, Abramovici H. Johanson-Blizzard syndrome: a
prenatal ultrasonographic diagnosis. Ultrasound Obstet
Gynaecol. 1999;13:450-2.
6. Townes PL, White MR. Identity of two
syndromes. Proteolytic, lipolytic and amyolytic deficiency
of exocrine pancreas and congenital anomalies. Am J Dis
Child. 1981;135:248-50.
7. Hurst JA, Baraitser M. Johanson-Blizzard
syndrome. J Med Genet. 1989;26:45-8.
8. Zenker M, Mayerle J, Lerch MM, Tagariello
A, Zerres K, Durie PR, et al. Deficiency of UBR1, a
ubiquitin ligase of the N-end rule pathway, causes
pancreatic dysfunction, malformations and mental retardation
(Johanson-Blizzard syndrome). Nat genet. 2005;37:1345-50.
9. Steinbach EJ, Hintz RL. Diabetes
mellitus and profound insulin resistance in Johanson-Blizzard
syndrome. J Pediatr Endocrinol Metab. 2000;13:1633-6.
|
|
|
 |
|
