|
|
|
Indian Pediatr 2010;47: 443-444 |
 |
Sanjad - Sakati Syndrome in a Neonate |
|
Kamalesh Pal
From Department of Pediatric Surgery, Maternity and
Children’s Hospital, AI Ahsa. Kingdom of Saudi Arabia.
Correspondence to: Dr Kamalesh Pal, PO Box 40129,
Consultant Pediatric Surgeon, King Fahad Hospital of the University
College of Medicine, King Faisal University, Al Khobar, 31952, Kingdom of
Saudi Arabia.
Email: [email protected]
Received: December 24, 2008;
Initial review: February 26, 2009;
Accepted: March 27, 2009.
|
|
Abstract
Congenital hypoparathyroidism, growth retardation and
dysmorphism is a rare autosomal recessive syndrome among Arab population
commonly known as Sanjad-Sakati syndrome(SSS).Several metabolic and
septic complications are known to manifest in the neonatal age. We
describe the first report of morbid pathological fractures affecting a
neonate with SSS.
Key words: Fracture, Neonate, Sanjad - Sakati syndrome.
|
|
S
anjad
Sakati Syndrome (SSS) is a rare but well known entity described mainly in
the Arab peninsula. The typical metabolic derangements lead to several
morbid manifestations commencing as early as neonatal period and include
hypocalcemia, seizures, nephro-calcinosis, increased susceptibility to
infections, stunted growth and mental retardation. This report highlights
an unusual occurrence of multiple longbone fractures in a neonate with SSS.
Case Report
A 34 week preterm boy (birthweight 1490 g) was born to
a 19 years old Saudi lady (G2P1) by
spontaneous vaginal delivery. Immediate neonatal period was uneventful.
Baby passed meconium and urine on first day and was started on feeds. He
developed fever and abnormal limb movements on 4th day associated with
vomiting and clonic seizures in all four limbs. The child had facial
dysmorphism in the form of microcephaly, deep set eyes, beaked nose,
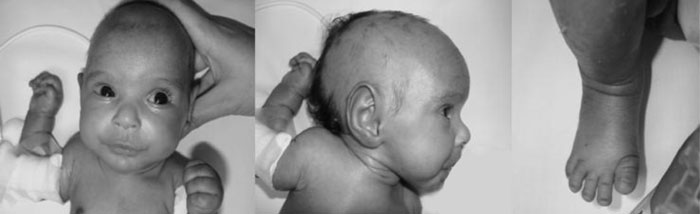
abnormal ear and micrognathia, short hands, and feet (Fig. 1).
Investigations revealed hypocalcemia (Ca=0.9mmol/L), hypomagnesemia
(Mg=0.45mmol/L); hypoparathyroidism (PTH=0.110 pmol/L; normal=1.59-6.89)
and hyper-phosphatemia (2.25mol/L; normal=0.81-1.58). Septic screen
revealed normal CSF, but blood culture at 17th day grew Klebsiella
sp. Baby was maintaining normal blood sugar and TORCH screening was
negative. He was started on parenteral calcium, magnesium, pheno-barbitone
and antibiotics. Feeding was regained on 20th day and baby started to
tolerate supplemented milk. On 22nd day, he was noticed to have multiple
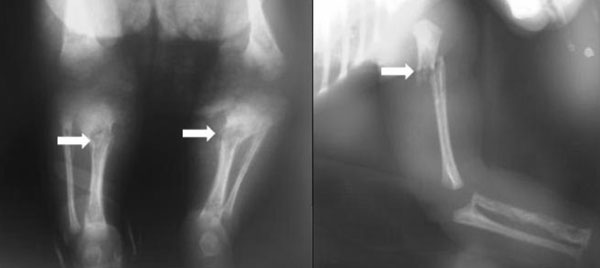
abscesses over shin with fractures of both tibia and left humerus (Fig.
2). Retrospectively, it was found that fracture sites correlated
with the sites attempted for IV placement by the NICU staff and a decision
to place jugular Hickman catheter was taken to prevent further
pathological fractures. A clinical impression of Sanjad-Sakati syndrome
was made due to classical dysmorphism, metabolic derangement, growth
retardation and seizures. Baby was put on enteral and parenteral calcium,
Vitamin D supplementation and pathological fractures took a long time (3
months) to heal.
 |
|
Fig. 1 Features of Sanjad
Sakati Syndrome (deep set eyes, abnormal ear, beaked nose,
microcephaly, short feet). |
 |
|
Fig.2 X-ray long bones
showing pathological fractures. |
Discussion
Sanjad Sakati Syndrome (SSS) or
hypopara-thyroidism-retardation-dysmorphism(HRD) syndrome is a rare but
well documented autosomal recessive syndrome predominantly seen in Arab
peninsula(1). It is characterized by congenital hypoparathyroidism (hypoPTH),
prenatal and postnatal growth retardation, seizures and a typical facial
dysmorphism, consisting of prominent forehead, deepset eyes, abnormal
external ears, microcephaly, microphthalmos, thinned upper lip, hooked
small nose, micrognathism, and small hands and feet. Metabolically, babies
suffer from often severe and fatal hypocalcemia, hypomagnesemia,
hyperphosphatemia and congenital permanent hypoPTH. These metabolic
derangements are responsible for nephrocalcinosis, medullary stenosis of
long bones and convulsions. Genetically this disorder has been mapped to
the long arm of chromosome 1 (1q42-q43). Mutations in the gene coding for
tubulin specific chaperone E (TBCE) have been identified as the cause of
the disease in Arabs. However, reports of a variant without TBCE mutation
has also been documented(2).
The phenomenon of multiple pathological fractures have
not been reported in SSS scenario so far. Although medullary stenosis due
to thickening of cortex in SSS has been documented, osteopenia and
pathological fracture in early neonatal age in SSS is intriguing. In
adults, PTH is considered anabolic to trabecular bone and catabolic to
cortical bone. Hypoparathyroidism leads to positive growth of cortical
bones and variable effects on trabecular bones usually measured by bone
densitometry(3). However, bone densitometry studies are deficient in
assesing mineral content and stress bearance aspect of bones carrying both
the trabecular and cortical types and this study was not conducted in our
child.
We conclude that pathological fractures could
complicate SSS in the neonatal period. Adequate mineral supplemented milk,
radiological survey of the skeleton and delicate handling of the limbs
particularly during lifting and placing IV access are some of the
precautions that may prevent such morbidity. Bone densitometry is
recommended to identify babies at risk of such complications.
References
1. Sanjad SA, Sakati NA, Abu-Osba YK. Kaddoura R,
Milner RDG. A new syndrome of congenital hypo-parathyroidism, severe
growth failure and dys-morphic features. Arch Dis Child 1991; 66:193-196.
2. Courtens W, Wuyts W, Poot M, Szuhai K, Wauters J,
Reyniers E, et al. Hypoparathyroidism – retardation – dysmorphism
syndrome in a girl : A new variant not caused by a TBCE mutation –
clinical report and review. Am J Med Genet 2006, 140: 611-617.
3. Duan Y, De Luca V, Seeman E. Parathyroid hormone
deficiency and excess; similar effects on trabecular bone but differing
effects on cortical bones. J Clin Endocr Metab 1999; 84: 718-722.
|
|
|
 |
|
