|
|
Brief Reports Indian Pediatrics 2004; 41:473-477 |
||||||
|
Differentiation of Fanconi Anemia from ‘Idiopathic’ Aplastic Anemia by Induced Chromosomal Breakage Study using Mitomycin-C (MMC) |
||||||
|
Rashmi Talwar†, V. P. Choudhry*, Kiran Kucheria From the Departments of Anatomy and Hematology*,
All India Institute of Medical Sciences, New Delhi 110 029, India.
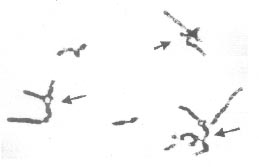
Key words: Aplastic anemia, Chromosomal breakage, Fanconi anemia, Mitomycin C. Aplastic anemia is an acquired disorder in more than 80% cases while the remaining 20% are inherited forms consisting of Fanconi anemia, dyskeratosis congenital and Schwachman syndrome. Of the acquired forms, ‘idiopathic’ aplastic anemia in which no cause is apparent accounts for approximately 65% of all cases of aplastic anemia(1). Fanconi anemia (FA) is a rare autosomal recessive disorder characterized by progres-sive pancytopenia, variable congenital anomalies, susceptibility to malignancies and induced chromosomal instability. However, a number of patients display only minor phenotypic variations or lack congenital abnormalities. Such patients present a diag-nostic dilemma for clinicians(2,3). FA Cells usually exhibit spontaneous chromosomal breaks and rearrangements that are amplified by the addition of DNA cross-linking agents like mitomycin C (MMC) and/or diepoxybutane (DEB). This hyper-sensitivity to the clastogenic effect of DNA crosslinking agents provides a unique marker for FA genotype and can be used as a diagnositic test(4). As the management of patients with FA differs from that of ‘idiopathic’ aplastic anemia, it is essential to differentiate these disorders at the earliest. In the present study, mitomycin C (MMC) was used to analyze induced chromosomal breakage in 29 patients with aplastic anemia with the aim to differentiate these cases. Subjects The study group included 29 patients (20 males and 9 females) with aplastic anemia with or without congenital malformations visiting the Hematology Clinic, AIIMS, New Delhi. The median age of patients at presentation in the present study was 8 years (range: 11 months to 35 years). Of the 29 patients, 27 were below 17 years of age while 2 patients (1 male and 1 female) were above 30 years of age. Family history was recorded in pre-designed proformas and a detailed pedigree was taken in each case. Informed consent was taken from the patients or their parents in case of minors. Induced chromosomal breakage study was carried out on peripheral blood samples obtained from all the 29 patients and 20 healthy control subjects. About 2-3 mL of venous blood was collected in heparinized syringe for each subject. Chromosomal spreads were obtained from PHA-stimulated 72 hours culture of peripheral blood lymphocytes using standard protocols. Cultures were set up in duplicates for mitomycin C (MMC)-stress test by adding MMC (40 ng/mg) at the time of initiation of cultures. Replicate sets of untreated cultures were set up from peripheral blood lymphocytes to serve as controls(5). A minimum of 50 metaphases (stained with Giemsa) was analyzed from each culture (MMC-treated, untreated and controls). The MMC-induced chromosomal breakages were then compared to those in the baseline (untreated) culture and healthy controls. Achromatic areas less than a chromosome width i.e., gaps were excluded in the calculation of chromosomal breakage frequency. Achromatic areas more than a chromosomal width were scored as breaks. Single chromatid breaks, isochromatid breaks and acentric fragments were scored as one break each while dicentric, ring chromosomes and chromosomal breaks were scored as two breaks each. Radial configurations were scored separately. The proportion of breaks and radial figures was expressed in percent, i.e. number of breaks or radial figures/number of mitotic figures × 100 (4-6). Results Induced chromosomal breakage study was successful in 24 out of 29 patients and the 20 healthy controls. Of these 24 successful cultures, mitomycin-stress test revealed a striking increase in both chromosomal breaks (Fig. 1) and chromatid exchange radial formations (Fig 2) in 10 patients (42%), slight increase in 9 patients (38%) and no induced chromosomal breakage in 5 patients (20%) when compared to controls. The mean breakage rate in FA patients was 750%, in ‘idiopathic’ aplastic anemia patients was 3.5% while that in healthy controls was 6%.
Ten patients with hematologic abnormalities diagnosed as Fanconi Anemia showed very high frequency of induced chromosomal breakage with almost every cell exhibiting multiple chromatid breaks and exchanges after exposure to MMC. Of these, 5 patients also manifested congenital mal-formations like skeletal anomalies (absence of radius and / or thumb), ectopic kidney, hypo- and hyperpigmentation of skin, café-au-lait spots, clinodactyly, polydactyly, microcephaly and growth retardation with failure to thrive. The hematologic abnormalities included aplastic anemia and thrombocytosis. Bleeding manifestations (bleeding from nose and gums) was also observed in 4 patients. Family history revealed death of siblings (with congenital anomalies) due to aplastic anemia in 3 patients of which one had consanguineous parents. Nine patients exhibited only slight increase in cells with chromosomal breakage as compared to baseline cultures. Four of these revealed two populations of lymphocytes, majority of cells (50-70%) treated with MMC appeared to have no chromosomal breakage while the remainder of cells in each case exhibited high number of breaks and exchanges characteristic of Fanconi Anemia patients. The mean breakage rate in these 4 patients was 120%, i.e., greater than controls (6%) and these were diagnosed as mosaics for Fanconi Anemia. The mean breakage rate of the aberrant cells was 520%. At presentation, 7 patients exhibited bleeding manifestations of which, 1 also had multiple congenital anomalies (hypopigmentation of skin, skeletal anomalies etc.). Two patients also had a history of death of siblings (with congenital anomalies) due to aplastic anemia. Of the remaining 5 patients with no induced chromosomal breakage, 3 patients had skin pigmentation problems and aplastic anemia while 2 had aplastic anemia only. Discussion Chromosome instability is a characteristic cytogenetic feature of a number of genetically determined human disorders collectively known as chromosome breakage syndromes. These disorders include Fanconi’s anemia (FA), Bloom’s syndrome (BS) and Ataxia telangiectasia (AT). In each of these, chromosome instability exists in the form of increased frequencies of breaks and inter-changes occurring either spontaneously or following treatment with various DNA-damaging agents(1,2). Although hypersensitivity to MMC and DEB in mitogen-stimulated peripheral blood lymphocyte cultures is accepted as a diagnostic criterion in FA, interpretation is difficult in mosaic patients(3). In the present study, 4 patients showed mixed populations of lymphocytes with hypersensitivity and normal sensitivity to MMC. Such a mosaicism has been reported earlier in 25-30% of FA patients(3,7). Aplastic anemia in the remaining 10 of the 24 patients with relatively no or slight induced chromosomal breakage could be due to other bone marrow failure syndromes or nutritional deficiencies. There is need to further evaluate these patients at the molecular level to screen for FA-like gene mutations(8). All the 14 Fanconi Anemia patients presented with hematologic abnormalities (pancytopenia and bone marrow aplasia) while only 6 patients (42%) had congenital anomalies (hypopigmentation of skin, skeletal anomalies, the most common being hypo-pigmentation of skin and clinodactyly). Eight patients also had bleeding manifes-tations mainly from nose and gums. MMC-stress test not only identified patients with congenital anomalies but also 8 patients (58%) who lacked any congenital malformations. Giampietro, et al.(9) reported 25-30% of FA patients without any congenital malformations all of which were diagnosed only after onset of hematologic abnormalities. A similar delay in diagnosis was observed in the present study that could be attributed to the lack of awareness among parents or referring physicians. In the present study, induced chromosomal breakage studies could diagnose Fanconi Anemia in 5 patients with history of affected siblings. Although these patients were diagnosed after the onset of aplastic anemia, MMC-stress test can be used to detect such patients even in the pre-anemic phase. This would help in avoiding drugs that are usually administered in acquired or ‘idiopathic’ aplastic anemia. Further, screening parents of FA patients can help detect ‘silent’ cases(9-11). A delay in diagnosis of FA can have serious consequences for patients and their families. An earlier diagnosis in FA patients (i.e., before onset of hematological abnormalities) could provide more time to find a suitable HLA compatible donor for bone marrow transplantation. Further, at-risk families (with an affected child) should be identified early and offered genetic counseling and prenatal diagnosis, as FA is an autosomal recessive disorder with a recurrence risk of 25%(5,10). Such a delay in identification of FA patients and at-risk families can be avoided by performing MMC-stress test in patients with macrocytosis and decreased platelet count as observed during screen- ing of complete blood count with differentials(11,12). Contributors: KK - cytogeneticist; designed and supervised the study. VPC - clinician; responsible for patient data and hematology. RT - conducted and analyzed cytogenetic tests. Funding: None. Competing interests: None stated.
| ||||||
|
References | ||||||
|
![]()