|
|
Indian Pediatrics 2003; 40:480-502 |
||||||||||||||||||||||||||||||||||||||||||||
|
The Fetal and Early Life Origins of Adult Disease |
||||||||||||||||||||||||||||||||||||||||||||
|
Caroline H.D. Fall
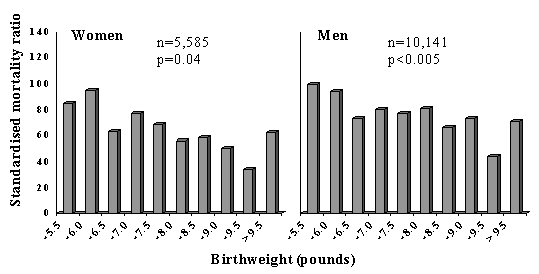
The fetal origins of adult disease (FOAD) hypothesis is based on the observation that men and women who were small at birth (low birthweight) have an increased risk of atherosclerotic cardiovascular disease (CVD) and the related diseases hypertension, type 2 diabetes and the Insulin Resistance Syndrome. Risk is increased further if they showed rapid weight gain in childhood or become obese. The hypothesis proposes that CVD is ‘programmed’ by under nutrition during critical periods of early development and that ‘poverty’ during early life creates a permanent vulnerability to ‘diseases of affluence’. This concept is arguably of greatest relevance to developing countries, where fetal growth restriction still affects large numbers of people, where economic progress is leading to the emergence of childhood and adult obesity, and where CVD and type 2 diabetes are rising rapidly. Its implication is that the prevention of adult disease should include strategies to improve maternal health and fetal growth. This paper reviews work leading to the FOAD hypothesis and the results of FOAD research in India. It also discusses some of the controversies surrounding the hypothesis, notably the debate as to whether the link between fetal growth restriction and adult CVD is mediated by environmental factors (such as maternal nutrition) or by genes. Key words: Adult disease, Fetal, Low birthweight, Obesity. Low Birthweight and Adult Cardiovascular Disease (CVD) The concept that adult health is influenced by events in early life is not new. In 1934, Kermack showed that death rates from all causes in the UK and Sweden fell between 1751 and 1930 with each successive year-of-birth cohort(1). He rejected one possible explanation, that babies were born healthier, and concluded that it was the result of better childhood living conditions brought about by social reforms. In 1977, Forsdahl discovered a geographical correlation within Norway between coronary heart disease (CHD) mortality in 1964-67 and infant mortality rates 70 years earlier (1896-1925)(2). He suggested that growing up in poverty caused ‘permanent damage’ perhaps due to a ‘nutritional deficit’, which resulted in ‘life-long vulnerability’ to an affluent adult lifestyle and high fat intakes. Studies in the UK a decade later shifted the focus back to pre-natal rather than childhood events when blood pressure was found to be inversely related to birthweight in young men and women in the 1946 national birth cohort(3). Barker showed that regional differences in stroke and CHD mortality in the UK in 1968-78 were predicted by neonatal mortality (a marker for low birthweight) in 1921-25(4). He went on to propose that the roots of cardiovascular disease lay in the effects of poverty on the mother resulting in undernutrition in fetal life and early infancy. Following up this theory, by using 1911-1930 birth records for one English county (Hertfordshire), Barker showed that lower birthweight and weight at one year were associated with an increased risk of death from CHD and stroke(5,6). There was an approximate doubling of CVD mortality from the highest to the lowest extremes of birthweight (Fig. 1), similar in men and women. Since then, other studies in the UK, Europe, and the USA have confirmed these findings(7-14). Studies using birth records with gestational age data indicate that it is restricted fetal growth rather than pre-term delivery which carries the risk of CVD(12). The effects are linear, graded across the whole range of birthweight (Fig. 1) and independent of adult socio-economic status(9,10,12). The majority of studies are limited to birthweight as a measure of fetal growth, but there is some evidence that body proportions at birth show stronger associations with CVD. For example low ponderal index (weight/length3) predicted CHD better than birthweight in Finland(11), and a low birthweight/head circumference ratio predicted stroke mortality in the UK(8).
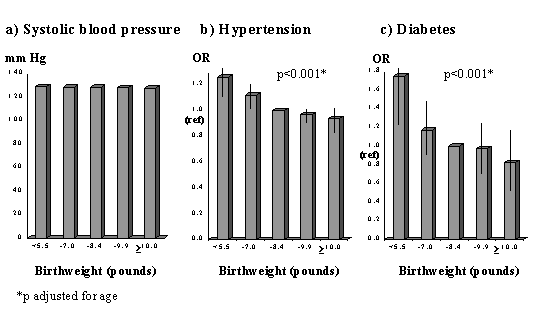
Fig. 1. Standard mortality ratios for cardiovascular disease in men and women born in Hertfordshire according to birthweight(6). CVD Risk Factors Subsequent work has shown that lower birthweight and other measures of small size at birth are also associated with higher levels of some ‘classical’ CVD risk factors. Insulin Resistance Syndrome: Blood pressure, type 2 diabetes, insulin resistance, and combinations of these (the insulin resistance syndrome (IRS)) are consistently related to low birthweight in a large number of studies in different populations(15-23). The strength of the association with the IRS (or Syndrome X) led Barker and Hales to suggest that it should be renamed the ‘small baby syndrome’(16). Studies relating blood pressure, diabetes and insulin resistance to birthweight, in adults and children, have been summarized in systematic reviews(24,25). It is notable that the associa-tions are stronger for disease outcomes, such as hypertension (Fig. 2)(26) or CVD, than for blood pressure measurements.
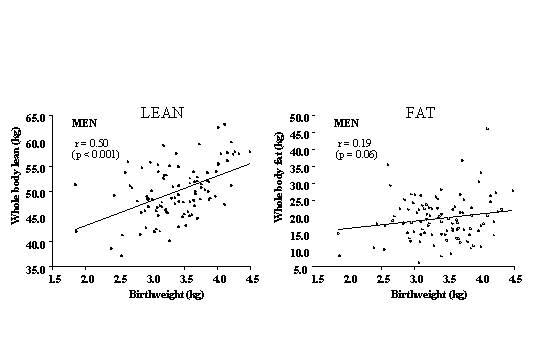
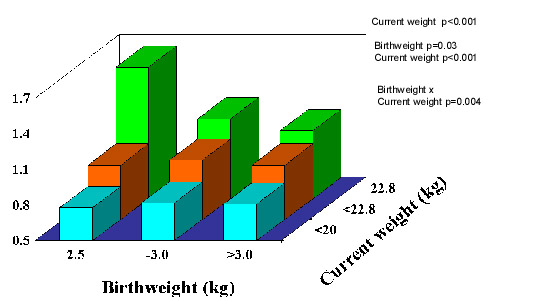
Fig. 2. Mean systolic blood pressure and odds ratios for hypertension and incident diabetes in 22,846 US men aged 48-83 years(26) Lipids and clotting factors: Although lipids show some associations with size at birth, these are weaker and less consistent. Total cholesterol shows a weak inverse association with birthweight(27). HDL-cholesterol con-centrations are positively, and triglyceride concentrations inversely, related to birth-weight in some(28) but not most studies(18,21-23,29,30). Post-prandial lipid concentrations, and Lp(a), fibrinogen and factor VII concentrations were unrelated to birthweight in Hertfordshire(31,32,28). Total- and LDL-cholesterol, apolipoprotein B and fibrinogen concentrations were associated with smaller abdominal circumference at birth, recorded in obstetric records in Sheffield, UK(33,34). PAI-1 was increased in low birthweight men in the one published study which has measured it(22). Cardiovascular Function: Arterial intima media thickness and carotid stenosis, examined using ultrasound, are increased in lower birthweight men and women(35,36) and flow-mediated dilatation, a measure of endothelial function, is reduced in young adults(37) and children(38) of lower birth-weight. In a study of microvascular function, post-ischemic recruitment of capillaries, and endothelium-dependent dilatation of pre-capillary arterioles were reduced in young adults of lower birthweight(39). Pulse wave velocity, a measure of poor arterial compliance, was increased in UK men and women with small head and abdominal circumferences at birth(40). Obesity: People who were heavier at birth tend to become ‘fatter’ adults as measured by body mass index(41). However, this may reflect increased lean mass rather than adiposity(42-44) (Fig. 3). There is no evi-dence that low birthweight leads to increased total body fat, but leptin concentrations were increased in low birthweight men and women in one study(45) and central obesity has been linked to small size at birth. The subscapular/triceps ratio is consistently higher in adults and children of lower birthweight(46,47,22). Waist circumference and waist/hip ratio are inversely related to birthweight in some studies(48,49,39) though this is less consistent(17).
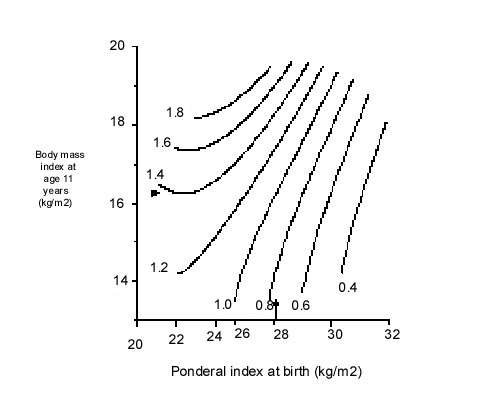
Fig. 3. Lean and fat mass measured by dual energy absorption (DEXA) in men aged 70-75 years, born in Sheffield, UK (n=102)(44) Post-natal Growth and Adult Obesity CVD and its risk factors are also linked to patterns of growth in infancy and childhood. In Hertfordshire, men with lower weight at the age of one year had increased CVD mortality(5) and type 2 diabetes(15). This was confirmed in men born in Finland(50). In Hertfordshire, men who had a low weight at one year also had higher fibrinogen(32) and cholesterol concentrations(29). Unlike weight gain during infancy, childhood weight gain is associated with an increased risk of later disease. Accelerated childhood weight gain (upward crossing of centiles) was associated with higher blood pressure in young adults in the UK(51). Similarly, in the Finland birth cohorts, although absolute weight and BMI in child-hood were unrelated to disease, an increase in BMI SD score between birth and 7 years was associated with an increased risk of CHD in men and type 2 diabetes in both sexes(50,52). Early adiposity rebound was also associated with increased adult diabetes; the cumulative incidence was 8.6% in men and women whose adiposity rebound occurred before the age of 5 years compared with 1.8% in those with an adiposity rebound after 7 years(53). What determines the age of adiposity rebound is unknown, but in Finland lower weight at one year predicted an earlier rebound(53). Increased height growth in childhood has been associated with CHD in women(13), and with blood pressure and insulin resistance in adults and children of both sexes(54,52,23). These measures of childhood growth often interact with measurements at birth in the prediction of adult disease. In Finland, an increase in BMI from birth to seven years was only associated with an increased risk of adult CHD in those who were small at birth(55) (Fig. 4).
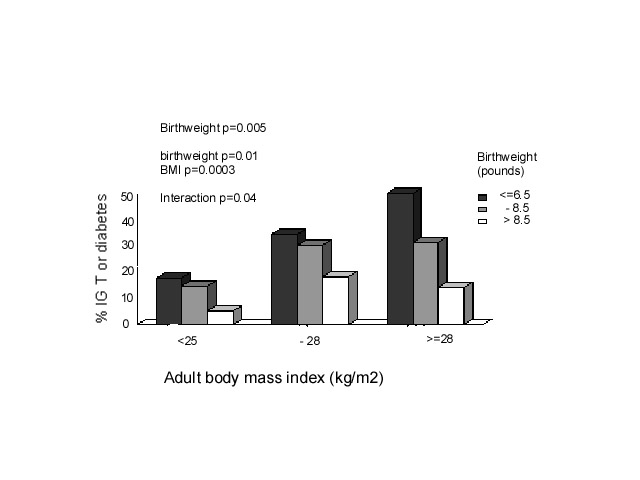
Fig. 4. Hazard ratios for death from CHD for men born in Helsinki 1924-33 according to ponderal index at birth and BMI at age 11 years55. Arrows indicate average values Adult obesity also adds to, and in some studies interacts with, the effects of low birthweight. The most adverse CVD risk profile is consistently found in men and women who were small at birth but who became obese adults (Fig. 5)(15). The effects of adult BMI on high blood pressure, type 2 diabetes, and insulin resistance are greater in individuals of low birthweight(15,9). The intriguing corollary of this is that high birthweight may protect against the adverse effects of adult obesity.
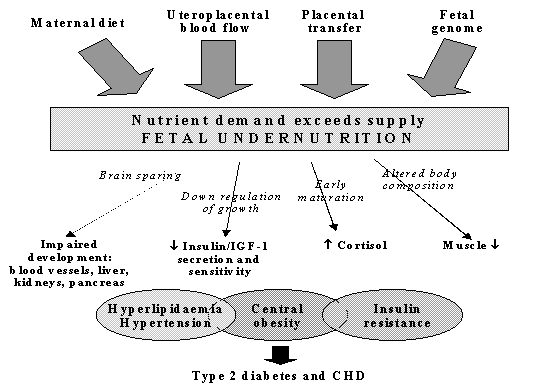
Fig. 5. Prevalence of impaired glucose tolerance (IGT) and Type 2 diabetes (%) in Hertfordshire men aged 60-71 (n=370)(15) The ‘Fetal Origins of Adult Disease (FOAD)’ Hypothesis Barker hypothesised that the associations between small size at birth or during infancy and later CVD reflect permanent effects of fetal undernutrition(56) (Fig. 6). The fetus is dependent on the transfer of nutrients from the mother and adapts to an inadequate nutrient supply in a number of ways: prioritisation of brain growth at the expense of other tissues such as the abdominal viscera, reduced secretion of/sensitivity to the fetal growth hormones insulin and IGF-I, and up-regulation of the hypothalamo-pitutary-adrenal (HPA) axis. The FOAD hypothesis proposes that although occurring in response to a transient phenomenon (fetal under-nutrition) these adaptations become perma-nent or ‘programmed’ because they occur during critical periods of early development.
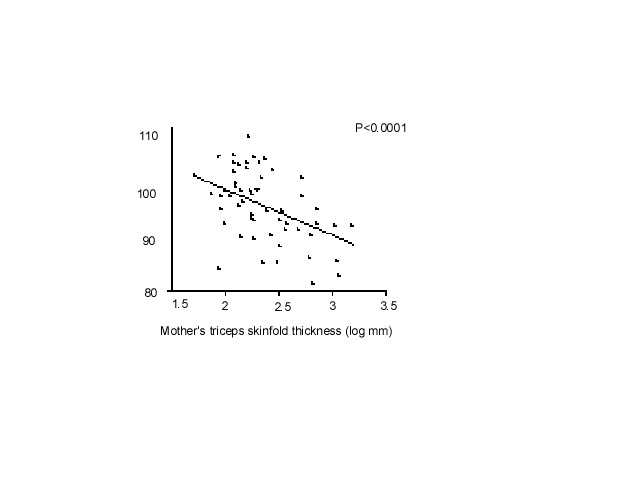
Fig. 6. The Fetal origins hypothesis The mechanisms by which this could occur at a cellular and tissue level have been reviewed(57,58). Programmed changes may include reduced insulin sensitivity(59), low muscle mass(60), pancreatic beta cell mass(61) and nephron numbers(62), altered arterial structure(63), and up-regulation of the HPA axis(64) and sympathetic nervous system(65) (Fig. 6). The FOAD hypothesis proposes that these changes also render the individual more susceptible to the effects of environmental stressors such as obesity arising in later life. The hypothesis is supported by examples in experimental animals of permanent structural and metabolic changes resulting from transient nutritional insults in utero. In rats, maternal protein restriction in pregnancy leads to higher blood pressure(66), impaired glucose tolerance(67), insulin resis-tance(68,69), and altered hepatic architecture and function(61) in the adult offspring. Animal experiments allow more sophisticated study of the mechanisms of programming at tissue and cellular level. For example, Hales and Ozanne have shown that insulin resistance in the adult offspring of protein-deprived rats results from reduced gene expression for PI3 kinase, an enzyme in the insulin-signalling pathway(67,68). There are a number of possible reasons why weight and height gain in childhood, on a background of fetal restriction, might be associated with disease. Low birthweight babies undergo compensatory post-natal growth, the rapidity of which may simply indicate the severity of the growth retardation(54). Alternatively rapid weight gain may be disadvantageous in itself(70), for example because of excess demand on tissues which are not capable of compensatory hyperplasia such as the pancreas(71,72), or through altered body composition. McCance observed excessive fat gain in pigs if they were placed on a high plane of nutrition after early post-natal undernutrition. He suggested that this emphasised the development of fat which maintains the capacity for growth throughout life, while muscle which loses the capacity for cell division, is unable to recover(73). Another possibility is that the hormones driving catch-up growth have adverse cardiovascular and metabolic effects(74). Low birthweight children who have caught up in weight and height have higher IGF-I concentrations, which in turn correlate with blood pressure(75). The Role of Nutrition Neonatal size is strongly related to maternal BMI, height, head circumference and even birthweight(76,77). This probably has both genetic and environmental components, but strongly suggests that the nutrition of a female throughout her life (during her own fetal life and childhood) as well as during pregnancy, influences the growth of her fetus. Nutritional effects on fetal growth are also shown by the drop in birthweight observed during famines(78). Fetal growth can be readily restricted in experimental animals by reducing maternal intakes of energy and protein during preg-nancy(79). On the other hand, randomised trials of energy and or protein supplements have shown only small effects on birth-weight(80). There is some evidence that improving the micronutrient quality of mothers’ diets leads to an increase in fetal growth, but this has not been well studied(81). Fetal growth depends on the uptake of nutrients at the end of a complex materno-fetal supply line(79). This includes not only the mother’s food intake but also her intermediary metabolism, endocrine status, cardiovascular adaptations to pregnancy (eg plasma volume expansion which influence uterine blood flow), and placental structure and function. The complexity of the link between maternal and fetal nutrition probably explains why the impact of maternal diet on fetal growth remains far from clear. If maternal nutritional status has important programming effects on the fetus, correlations would be expected between measures of maternal body composition and diet and CVD or its risk factors in the offspring. The problem is that there are limited data to test this, especially in populations old enough to measure disease outcomes. What data exist are limited to maternal measurements recorded in clinical obstetric notes (usually only weight and height), old dietary surveys, and famine studies. Low maternal weight gain, BMI or skinfolds in pregnancy are associated with higher offspring blood pressure(82,83) (Fig. 7) and insulin resistance(20,23). Conversely, in Finland, mortality from CHD was increased in men and women whose mothers were short and had a high BMI(11). Among men and women born during the Dutch famine of 1944-45, late gestation exposure to famine was associated with glucose intolerance, insulin resistance, and a (small) increase in type 2 diabetes(84). Early gestation exposure was associated with higher LDL/HDL cholesterol concentrations and (in women) higher BMI and waist circumference(85). Three recent studies suggested that the balance of maternal protein and carbohydrate intakes during pregnancy is related to blood pressure in the offspring(86,87,83).
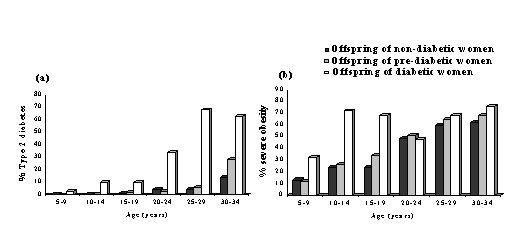
Fig. 7. Children’s systolic pressure adjusted for sex and current weight according to the mother’s triceps skinfold thickness at 15 weeks of gestation(82) All these studies had only crude dietary data and large losses to follow-up, and it is difficult to put the findings together. A number of prospective observational studies of maternal diet in pregnancy are due to report outcomes in the children during the next 2-3 years, including the Princess Anne Study(88) and Southampton Womens’ Survey in the UK(89) and the Pune Maternal Nutrition Study in India(90). These have collected good data on maternal body composition and diet, followed up the cohorts of children, and will determine whether maternal diet has important effects on CVD risk in the offspring. If it does, this clearly has strong public health implications. Maternal Diabetes and Fetal Macrosomia Although the main focus of this paper is fetal growth restriction, recent data show that maternal diabetes, which results in fetal ‘overnutrition’ and macrosomia, also has adverse long-term effects. Offspring of diabetic mothers have an increased risk of obesity and type 2 diabetes compared with offspring of non-diabetic mothers or women who became diabetic after the pregnancy (Fig. 8)(91). The difference in risk holds true for siblings born before and after the onset of maternal diabetes and is not seen among offspring of diabetic fathers(92). Gestational diabetes produces a U-shaped or J-shaped relationship between birthweight and adult type 2 diabetes(93,94). With increasing levels of obesity worldwide, gestational diabetes is also increasing(95) and may make an important contribution to the incidence of type 2 diabetes.
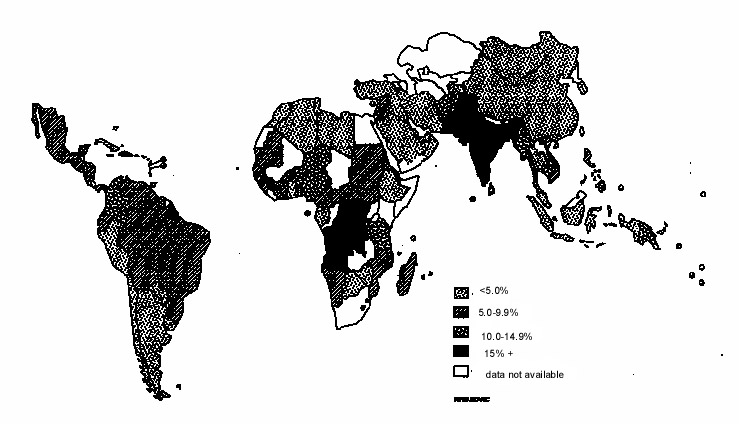
Fig 8: Prevalence of (a) type 2 diabetes and (b) obesity in offspring of non-diabetic, pre-diabetic and diabetic women (Pima Indians)(91) Relevance of FOAD to India and Developing Countries The linear and graded trends in CVD mortality with birthweight (Fig. 1) suggest that the majority of the world’s population experience sub-optimal fetal growth. Rates of intra-uterine growth retardation are highest in developing countries. In India the mean full-term birthweight is 2.6-2.7 kg, almost 1 kg lower than in Western Europe(96). A world map of intra-uterine growth retardation highlights South Asia as one of the worst-affected regions (Fig. 9)(97), with an estimated 25% of full term babies born weighing less than 2.5 kg. A high proportion of infants and children in India are still undernourished(98), but with economic progress childhood and adult obesity is an emerging problem, especially in cities(99). It is estimated that 20% of women and 16% of men in India will be overweight (BMI>25 kg/m 2) by the year 2020(99). Furthermore, there is evidence that at any level of BMI, South Asian men and women have a higher fat mass, more centrally distributed fat, and a higher risk of obesity-associated disease than white Caucasians(100).
According to the FOAD hypothesis, increasing child and adult obesity in combination with persistently poor fetal growth creates a high risk for adult CVD and diabetes. There is certainly an epidemic already unfolding in India and other countries undergoing rapid economic development and modernization(101). The projections are extremely frightening. In India, mortality from cardiovascular disease is expected to rise by about 60%, and overtake deaths from infectious disease, by 2015-20(102). The prevalence of type 2 diabetes has increased by 40% in Chennai, India between 1988 and 1994(103). King has predicted(104) that the prevalence of type 2 diabetes will rise by 30% worldwide, from 4.0% to 5.4% by 2025, and that the proportional rise will be greatest in developing countries (48%), especially China (68%) and India (59%). India will have more people with diabetes (≈ 57 million) than any other country, with the greatest numbers in the 45-64 year age group. It is also likely that type 2 diabetes will start to emerge in children. What evidence is there that poor fetal growth makes a contribution to India’s high rates of CVD and diabetes? The first study to examine this question was the Pune Children’s Study, in which CVD risk factors were measured in 201 children born in the KEM Hospital in Pune(105). Birthweights were obtained from labour ward records. When they were initially studied, at the age of 4 years, lower birthweight children had higher plasma insulin and glucose concentrations after an oral glucose load and higher IGF-I concentrations, but no other evidence of an adverse CVD risk profile(105,106). The study sample was expanded and the children were restudied at 8 years, when there were clear inverse relationships between birthweight and a number of features of the insulin resistance syndrome: systolic blood pressure, insulin resistance, total and LDL-cholesterol concentrations, and subscapular-triceps fat ratio(107). The most adverse risk profile was in children who were small at birth but had a high weight and fat mass at 8 years (Fig. 10). There was no diabetes at this age, and it is not yet known if insulin resistance in childhood persists into adult life or predicts future type 2 diabetes. However, these data suggest that a higher fat mass in children is associated with insulin resistance, and that this is further exacerbated by low birthweight.
Fig. 10. Relative insulin resistance (HOMA) in 8-year-old
children, Pune, India (n = 477)(107).
TABLE I 2-hour plasma glucose concentrations and other components of the insulin resistance syndrome according to birthweight in men and women aged 20-45 years in Mysore
p value adjusted for age, sex, and body mass index.
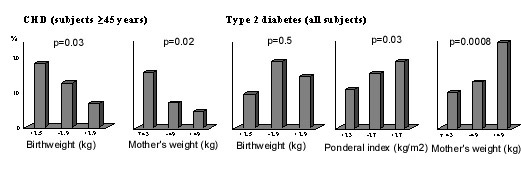
Further evidence came from studies carried out at the Holdsworth Memorial Hospital (HMH) in Mysore, South India. Built in 1905, HMH has preserved its obstetric notes, recording the birthweight, length and head circumference for all babies born in the hospital, since 1934. During 1993-1995, over 500 men and women born in HMH and still living in Mysore were traced by a door-to-door survey of central Mysore and matched to their birth records(108). Among men and women aged over 45 years, the prevalence of coronary heart disease (CHD, defined using ECG changes and an angina questionnaire) was 11% overall, and as in the Western studies, higher in those of lower birthweight (Fig. 11). It was also higher in those whose mothers were below average weight at antenatal booking. Lower birthweight was also associated with poorer adult lung function (FEV1 and FVC)(109).
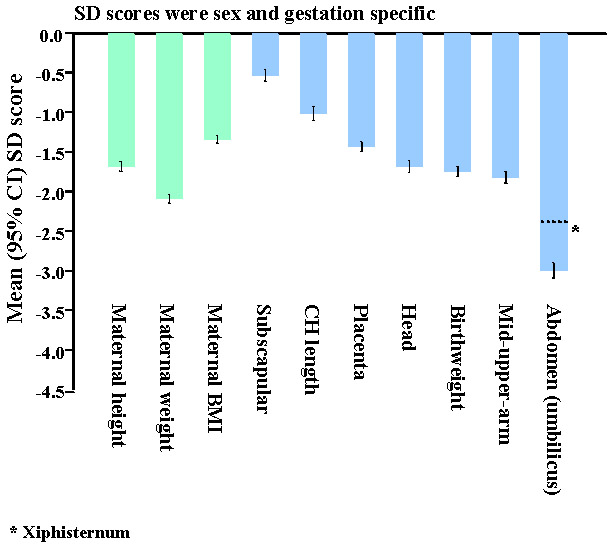
Fig. 11. Prevalence (%) of coronary heart disease and type 2 diabetes in men and women aged 38-60 years in Mysore(108,110). The findings for diabetes were different(110). Although lower birthweight was associated with higher fasting insulin concentrations, it was not associated with higher rates of type 2 diabetes (Fig. 11). The prevalence of diabetes was increased in men and women who were short but relatively heavy at birth, with a high ponderal index (birthweight/birthlengths). They were characterised by a low 30-minute insulin increment, a marker of reduced first phase insulin secretion. The prevalence of diabetes was also increased in offspring of mothers with a higher body weight (Fig. 11) and larger external pelvic diameters. The authors speculated that maternal glucose intolerance may explain these findings(110). Since that first Mysore study, the birth records have been used to trace more people, including younger adults. Interestingly, their data were more akin to Western populations. Lower birthweight was associated with higher two-hour glucose concentrations, higher rates of type 2 diabetes and impaired glucose tolerance, and higher serum triglyceride concentrations (Table 1). In marked contrast to a large number of studies around the world, there was no association in either Mysore cohort between birthweight and blood pressure(111). The problem with retrospective studies like the ones in Pune and Mysore is that only a small percentage of the original births could be followed up (6% in the first Mysore study): those who survived to adulthood, stayed near their place of birth, were successfully traced, and agreed to take part in the research. In addition, gestational age data as recorded in routine clinical notes, was incomplete and unreliable. Two prospective studies based on large birth cohorts are currently under way in New Delhi and Vellore, and are due to report their findings this year(112,113). These studies, originally set up by ICMR, each include more than 1,000 young adults born during 1969-1973 in both rural and urban settings. They have good quality data on maternal pre-pregnant weight and height, gestational age at delivery, neonatal anthropometry measured by trained researchers, growth data through infancy, childhood and adolescence, high rates of adult follow-up and information on adult lifestyle. These studies will clarify the relative importance, in Indian populations, of maternal size and body composition, and an individual’s own neonatal size, childhood growth, and adult body composition and lifestyle to CVD risk. In the meantime, contemporary birth cohorts are being followed up to examine effects of maternal body composition, diet and metabolism on CVD risk in Indian children. The Pune Maternal Nutrition Study (PMNS) is a prospective observational study of maternal diet and offspring outcome among 800 rural Indian women(90,114). Detailed anthropometry of the PMNS babies at birth showed that their body composition differed from UK babies (Fig. 12). They were lighter by almost 2 standard deviations (SDs), and non-fat soft tissues such as muscle (mid-upper-arm circumference) and abdominal wall and viscera (abdominal circumference) showed a similar deficit. Measurements of fat, however, were substantially ‘spared’ (-0.5 SD); although small and thin, they were relatively adipose. Hence, the adiposity of adults in India, which is thought to at least partly explain their high risk of diabetes and CVD, is present even at birth. This was confirmed in urban Pune babies(115) and in babies born in Mysore(116). The Mysore study showed that adiposity was further increased in babies born to mothers with gestational diabetes, the incidence of which was high (6%)(116).
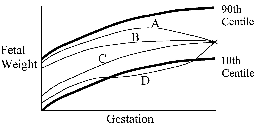
Fig. 12. SD scores for maternal and neonatal measurements in Pune compared to Southampton, UK(114). The PMNS showed that although there were no associations between neonatal size and the mother’s energy or protein intakes, her intake of milk at 18 weeks and of green leafy vegetables (GLVs) and fruits at 28 weeks were directly related to the babies’ size(90). Women eating GLVs every alternate day or more had babies almost 200 g heavier, than those who never ate them (OR for low birthweight: 0.43 (95% CI 0.12 to 0.99). All birth measurements (including body fat) were increased. Higher intakes were also associated with higher maternal fasting plasma insulin and triglyceride concentrations, and lower albumin concentrations, all of which were related to increased neonatal size. Controversies for the FOAD Hypothesis; Clinical Importance It has been argued that the increased risk of adult disease attributable to intra-uterine undernutrition is very small(117-119). Statistically birthweight explains little of the variation in adult disease. Effects are most marked at the extremes of birthweight, where there are relatively few individuals. Based on the Hertfordshire data Joseph estimated that 26% of CHD deaths would be averted if all babies weighed 9-9.5 pounds at birth, 9% if babies were born one pound heavier, and only 2% if more realistic increases in birthweight (100-200g) were achieved(117). Boyko calculated a larger effect: a type 2 diabetes population attributable fraction (PAF) for low birthweight ranging from 0.02 to 0.25 in 4 studies, compared with PAFs for adult BMI of 0.25 to 0.43 based on NHANES data(118). These calculations are potentially mis-leading. Crude birth measurements and imprecise estimates of gestational age at birth would lead to under-estimation of effects. Birthweight is an insensitive marker of the dynamic process of fetal growth(79) (Fig. 13), and does not capture the effects of fetal undernutrition on body composition or the development of specific tissues. Babies born after exposure to the Dutch Famine during early gestation had normal birthweights, and yet an increased risk of adult obesity and dyslipidemia(85). The large numbers of men and women required to show the effect of birthweight on CVD (Fig. 1) contrast with the small numbers of rats required, for example, to show an effect of nutritionally-induced defects in gene expression in the insulin signalling pathway on adult glucose intolerance(68). The true impact of intra-uterine undernutrition will not be known until there are better tissue-level markers of programming than birthweight.
A fetus can reach a given birthweight via a variety of possible different growth trajectories. These fetuses may be a different body composition, and may have differing disease risk in adulthood. A Late growth restriction and fetal wasting. B Early growth restriction. C Normal growth. D Late growth restriction followed by catch-up growth. Fig. 13. Possible fetal growth trajectories(79).
In addition, effects of size at birth are conditioned by childhood growth and adult obesity. Data from Finland, show that combinations of birth, infant and childhood measurements predict large differences in the risk of CHD, hypertension and diabetes (Fig. 4)(51,55,120). Controversies for the FOAD Hypothesis; Genes ‘ Versus’ Environment The fetal origins hypothesis has also been called the ‘thrifty phenotype’ hypothesis, a name coined by Hales and Barker after Neel’s ‘thrifty genotype’(121,122). ‘Thrift’ can mean ‘saving’ (storing resources in times of plenty in preparation for leaner times) or ‘economy’ (making judicious use of meagre resources). Neel suggested that diabetes is caused by ‘thrifty’ genes which were selected for in mankind’s distant past when the supply of food was precarious. He suggested that these genes conveyed a ‘fast insulin trigger’ and thus the ability to store food rapidly as fat (savings), which became diabetogenic in a modern setting of plentiful nutrition. The thrifty phenotype hypothesis, on the other hand, suggests that the undernourished fetus develops insulin resistance and other metabolic changes as a strategy for immediate survival, to down-regulate and prioritise growth (economy), for which it pays a price later in life, generally after the reproductive period. Both thrifty genes and the thrifty phenotype could become detrimental on exposure to plentiful nutrition. Variations on the thrifty genotype hypothesis are the strongest contenders to the FOAD hypothesis as an explanation for the associations between low birth weight and CVD risk. Correlations between parent and offspring birthweights and between birthweights of half-siblings related through either the mother or the father show stronger maternal than paternal effects, which suggests that the ‘maternal environment’ is a more powerful influence on fetal growth than genes (123,124). The birthweight of babies born after ovum donation are strongly related to the weight of the recipient mother, but not that of the donor mother(125). Nevertheless there are significant genetic effects on size at birth(77,126). Hattersley proposed that, since insulin is a major growth hormone in fetal life, genes associated with either insulin resistance or reduced insulin secretion would lead to reduced fetal growth as well as an increased risk of adult diabetes (the ‘fetal insulin hypothesis’)(127). That this is biologically feasible is shown by the fact that birthweight is reduced in a number of genetic syndromes causing impaired insulin secretion or insulin resistance(126). These are too rare to explain the observed birthweight-disease associations, but more commonly occurring polymorphisms, linked both to small size at birth and adult diabetes, have recently been described(128-130). The robustness of these associations remains to be tested; they have not been replicated in other studies(131,132). Twin studies have classically been used to distinguish between genetic and environmental effects. Twins are smaller at birth than singletons but do not experience greater cardiovascular morbidity or mortality. Studies linking CVD risk to the difference in birthweight within twin pairs have shown inconsistent results. For example, although a study using the Danish twin registry showed that the smaller of monozygous twin pairs was more likely to become diabetic(133), this has not been confirmed in other twin populations(134). The mechanisms under-lying the growth restriction of twins probably differ from those limiting growth in singleton fetuses. Higher disease concordance rates for monozygous than dizygous twins may reflect their shared intra-uterine environment as well as shared genes. Twin-twin interactions, for example the diffusion of steroid hormones from one twin to another, may reduce within-pair differences in programming effects. These features of the biology of fetal growth in multiple pregnancies limit the conclusions that can be drawn, in relation to the FOAD hypothesis, from twin studies(135). Recent reports of associations between low offspring birthweight and an increased risk of CVD and insulin resistance in parents (both mothers and fathers) could be evidence of common genetic factors(136-139). However, intergenerational effects may also have environmental explanations. Assortive mating and shared lifestyle (socio-economic status, nutrition, smoking, and stress) could lead to both low birthweight and later CVD in both parents. Some but not all of these were taken into account in these studies. Associations shown between low offspring birthweight and type 2 diabetes in fathers but not mothers is more powerful evidence of genetic effects(140,141). The lack of effect in mothers argues against these resulting from a shared environment. Maternal gene effects may have been masked by gestational diabetes or the effect could act entirely through paternal genes (paternal imprinting). Fathers of low birthweight babies did not, however, have increased insulin resistance or diabetes in two other recent studies from India(142,143). The time trends in CVD and type 2 diabetes in western countries and in different socioeconomic groups during the 20th century, and the recent rise in developing countries, suggest a susceptibility to environmental changes, which could either have a genetic basis (thrifty genotype) or arise from fetal programming (thrifty phenotype). However these would make different predictions for the future. The former would predict continuing high levels of disease unless people reduce their lifestyle risk factors and become less obese. The thrifty phenotype hypothesis would predict a downturn in disease as better nutrition of girls and mothers leads to improved fetal nutrition. CHD has been falling in the USA and Europe for 30 years despite increasing adult obesity, and only modest reductions in classical lifestyle-related risk factors. The incidence of stroke has also fallen since the early 1950’s in the UK. In contrast, type 2 diabetes is increasing in all populations worldwide. However, the increase has been less marked in developed than developing countries(104), and a fall in incidence has been reported in one population, the Nauruan islanders(144). The ‘genes versus environment’ debate is currently stimulating a great deal of hypothesis-testing research in this field. With increasing understanding of epigenetic effects and gene-environment interactions, it is no longer possible to think of diseases as being either ‘genetic’ or ‘environmental’(145). It was recently shown that an allele of the PPAR- r gamma gene is associated with increased insulin resistance, but only in men and women of low birthweight(146). It is clearly possible to permanently alter gene expression by manipulation of intra-uterine nutrition(68). Such epigenetic effects may persist across generations. For example, feeding ‘agouti’ mice with a methyl-supplemented diet during pregnancy leads to permanent and heritable effects on offspring coat colour, which is regulated by well-characterised genes(147). Imprinted genes, several of which play a role in fetal growth, are thought to be particularly susceptible to epigenetic effects. At this time, it seems likely that environmental effects, genes and interactions between the two contribute to the observed associations linking birthweight to adult disease.Public Health Implications and Future Research The FOAD hypothesis is attractive because it suggests that several common degenerative diseases could be prevented by improving maternal health and fetal development. Although it is not yet clear what optimal fetal growth is and how it can be achieved, it may turn out to be more attainable than persuading men and women in middle age to return to ‘primitive’ lifestyles, and more positive than concluding that large numbers of people have genes that are incompatible with modern living. Data from experimental animals provide powerful evidence that a mother’s nutrition programs the metabolism of her offspring. However, there is still insufficient evidence that maternal nutrition underlies cardiovascular disease in humans, and insufficient data to make specific dietary recommendations to pregnant mothers. Future research should focus on the determinants of fetal growth, including maternal diet, and incorporate follow up of infants and children for cardiovascular disease-related outcomes. Epidemiological research should go hand in hand with studies of the genes known to influence fetal growth and those associated with cardiovascular disease, and gene-environment interactions. There is no evidence that the current pediatric policy of encouraging catch-up growth during infancy in low birthweight babies is harmful. However, prevention and treatment of obesity in childhood should become a public health priority. Current data suggest that the greatest benefit in terms of future risk-reduction will be in low birthweight individuals. Further research is needed into effective interventions to prevent and treat obesity. Women intending to become mothers should avoid obesity, which is major cause of gestational diabetes. The optimal range of maternal body mass index, and the relative disadvantages of excessive thinness and ex-cessive adiposity, are, however, still un-known. Dietary supplementation of under-nourished mothers may have the effect of simply making them more adipose, which may have disadvantages for the fetus; this needs to studied further. Acknowledgements I would like to thank the following with whom I have had the pleasure of collaborating in research for the past 12 years: Dr. Anand Pandit, Dr. Chittaranjan Yajnik, Dr. Kurus Coyaji and Dr. Sheila Bhave, KEM Hospital, Pune; Dr. BDR Paul, Dr. Prasad Karat, Dr. K Kumaran, Dr. SR Veena and Dr. GV Krishnaveni, Holdsworth Memorial Hospital, Mysore; Dr. Santosh Bhargava, Sunderlal Jain Hospital, New Delhi; Dr. HPS Sachdev, Maulana Azad Medical College, New Delhi; Dr. KS Reddy & Dr. R Lakshmi, All India Institute of Medical Sciences, New Delhi; Dr. P Raghupathy, Dr. J Richard and Dr. B Antonisamy, CMC Hospital, Vellore and Dr. Ramesh Potdar, Jeevak Hospital, Mumbai. The research has been supported by the Medical Research Council, the Wellcome Trust, the Parthenon Trust, the Department for International Development, and the Wessex Medical Trust. I acknowledge the support of Sneha-India (Society for Natal Effects on Health in Adults; www.sneha-india.org).
| ||||||||||||||||||||||||||||||||||||||||||||
|
References | ||||||||||||||||||||||||||||||||||||||||||||
|
|
![]()