|
|
Brief Reports Indian Pediatrics 2000;37: 526-531 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Hemophagocytic
Lymphohistiocytosis : A
Case Series
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Leni
Grace Mathew
Thomas Cherian Annie Sudarshanam* Ipeson Korah# Shyam Kumar N.K.# P. Raghupathy
Hemophagocytic lymphohistiocytosis (HLH) is a rare fatal disease of infancy and early childhood(1). It is a disorder of the mononuclear phagocyte system, characterized by proliferation and activation of benign histiocytes, causing dysfunction of various organs(2). Two forms of HLH have been described; primary or familial (autosomal recessive) hemophagocytic lymphohistiocytosis (FHL) and secondary or sporadic. The sporadic variety of HLH is often associated with infections or malignancy(3). Untreated HLH has a very high mortality rate. Immuno-chemotherapy followed by allogenic BMT offers hope for long term survival in these patients(3). Since this form of therapy is currently available in our country(4), early diagnosis of HLH is important so that appro-priate treatment can be initiated. We report a series of cases of HLH diagnosed in our hospital over a period of seven years. This report along with previously published reports document that HLH is prevalent in India(5,6).
Clinical records of children aged 0-12 years, admitted to one of the two Child Health Units of our hospital with a diagnosis of HLH during the period 1991 to 1997, were reviewed. The diagnosis of HLH was based on the criteria laid down by the FHL study group of the Histiocytic Society(7). The clinical criteria included fever >38ºC. lasting for >7 days and splenomegaly. The laboratory criteria were cytopenia affecting at least two of three lineages in the peripheral blood (Hb <9 g/dl, Platelets <100,000/cu mm or ANC <1000/cu mm), hypertriglyceridemia and/or hypofibrinogenemia. The histological criteria included demonstration of hemo-phagocytes in the bone marrow, spleen or lymph node and absence of marrow hyperplasia or malignancy. Diagnosis of FHL required a family history of HLH. The data collected included details of family history, parental consanguinity, clinical and laboratory features, treatment and outcome.
Clinical
Features The interval between onset of symptoms and diagnosis of HLH varied from eight days to four months. Fever more than seven days duration and hepatosplenomegaly were present in all patients (Table I).
Laboratory
Features
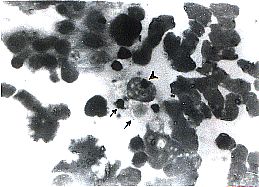
Histological
Characteristics
Treatment
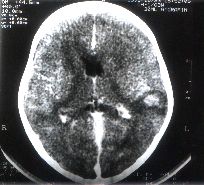
and Outcome The time interval between diagnosis and death ranged from two days to six months. Of the four patients who died in the hospital, death occurred within 10 days of diagnosis in three. The cause of death in these patients were Pseudomonas aeruginosa septicemia in one, profuse bleeding following liver biopsy in another and the disease itself in the third patient. The fourth patient lived for six months after the diagnosis of HLH was made. During this period he was admitted five times with recurrence of fever, hepatosplenomegaly and cytopenia. He improved with supportive therapy initially. In view of his recurrent symptoms, during the second relapse he was started on intravenous immunoglobin (IVIG). Following this fever subsided, hepatosplenomegaly regressed and cytopenia improved. However, he returned two months later with recurrence of symptoms. Since he had previously responded to supportive therapy and IVIG, chemotherapy was deferred. Six months after diagnosis, he developed generalized seizures with altered sensorium along with fever. CSF examination revealed 26 leukocytes/cu mm with 95% lymphocytes, 40 RBC/cu mm, sugar 51 mg/dl and protein 90 mg/dl. There were no malignant cells or hemophagocytes on the cytospin smear of CSF and bacterial culture was sterile. CT scan of the brain showed a contrast-enhancing lesion in the temporo-parietal cortex (Fig. 2). He was treated with etoposide 150 mg/m2/ dose and dexamethasone 3 mg/kg/day but succumbed to the illness three days after the first dose of chemotherapy. This patient’s sibling who died of HLH with CNS involve-ment (in another hospital) had a similar lesion in the cerebellum on his CT scan. None of the other patients were given chemotherapy because three of them died by the time the diagnosis of HLH was made and the fourth patient was discharged at parents’ request. The remaining two patients who had documented bacterial infection recovered completely with appropriate antibiotic therapy. Table I: Clinical Features at Admission
Table II: Laboratory Features at Admission
The aim of this article is to describe the clinical and laboratory features of HLH, to document the occurrence of this condition in India and to sensitize pediatricians to this problem in order to facilitate early diagnosis and treatment. The clinical features at presentation of our patients were similar to other series(3,8,9). Three patients had seizures at the onset of symptoms, but none of them had hemo-phagocytes in the CSF. One patient developed CNS involvement later during the course of the illness and succumbed to it. Involvement of CNS commonly occurs during the course of the illness as in our patient. However, early and predominant cerebro-meningeal involvement in HLH is well known(10,11). The other organs, which can be involved, are lungs, thymus, heart, kidneys, intestine and pancreas(3). None of our patients had histo-logically documented involvement of any of these organs, though one patient had hematemesis. The diagnosis of HLH is often difficult due to the rarity of the disease and lack of a specific laboratory test, resulting in under diagnosis. In an attempt to overcome these difficulties, the FHL study group of the Histiocytic Society has proposed diagnostic guidelines for HLH(7). However, it is important to know that some patients may present with an incomplete picture and develop the remaining abnormalities later. There are also less well-known presentations such as early cerebro meningeal involvement, chronic persistent hepatitis or neonatal or prenatal presentation(3). In the absence of any specific marker, a strong clinical suspicion is often warranted for early diagnosis of HLH. Langerhans Cell Histiocytosis (LCH) and malignant histiocytosis are clinically and histologically distinct from HLH. Without treatment, FHL is usually rapidly fatal, with a median survival of about two months(12,13). Combined use of steroids and epipodophyllotoxins has prolonged the short-term survival. Recently, immunotherapy with cyclosporin and ATG has been shown to be effective(14). A major therapeutic breakthrough was achieved with allogenic BMT(15). The current recommendation is chemotherapy followed by BMT(3). Sporadic HLH carries a better prognosis. IAHS triggered by bacterial infection is associated with a high recovery rate, but the outcome of EBV associated HLH is poor(16). Contributors: LGM, TC and PR were involved in the diagnosis and management of these patients and in the preparation of the manuscript. As provided laboratory assistance. IK and SK reported the radiographs.
Funding:
None.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
![]()